

Acceso a CCPS
No, ni tampoco para enviar las comunicaciones. El sistema de envío de comunicaciones se realiza mediante un código de referencia que remite el sistema por correo electrónico, para garantizar la identidad de la persona que está realizando la comunicación. Lo único imprescindible es disponer de una cuenta de correo electrónico y mantener ésta permanentemente actualizada en los datos de contacto de la aplicación. La dirección de correo electrónico a la que se envía el código de referencia es únicamente el de la persona titular de la clave de acceso. Para recibir los códigos de referencia y hacer los envíos cómodamente, se recomienda mantener abierta la aplicación del correo electrónico de su ordenador desde la cuenta de correo de la persona titular de la clave de acceso.
Cualquiera de última generación (Mozilla, Chrome, Edge, Safari, etc.), nunca Internet Explorer.
Introduzca en su navegador la siguiente URL: https://ccps.aemps.es/ccps/faces/login.xhtml. Aparecerá la pantalla principal de la aplicación con los campos etiquetados como “Usuario” y “Contraseña”. Si dispone de claves de la antigua aplicación PMPS, introdúzcalas, son igualmente válidas. Si no tiene o desea más claves de usuario para la misma empresa (esta aplicación permite tener varios usuarios para la misma cuenta de la empresa), pinche el enlace “Solicite alta en CCPS”. Pulse el botón “Enviar” y accederá a las opciones de la aplicación. Puede consultar el manual de alta de usuarios que se descarga en la portada de la aplicación: https://sede.aemps.gob.es/PSCH/PS/docs/CCPS-alta.pdf
El nombre de usuario no es sensible a las mayúsculas y minúsculas, por lo que podrá escribirlo como desee. Sin embargo, la contraseña sí lo es, por lo que ésta tendrá que escribirse exactamente igual que como se le ha proporcionado, o como usted la escribió la última vez que la cambió.
Sí, basta con rellenar el formulario que aparece al pinchar “Solicitar alta en CCPS”.
Los correos de la aplicación se recibirán tanto en la dirección correspondiente a los datos de empresa comunicante, como en los de los usuarios de esa misma empresa que aparezcan en los datos generales de cada comunicación. Sin embargo, la clave de referencia necesaria para el envío de documentación, por seguridad, solo se enviará a la dirección de correo que se indicó cuando se solicitó la clave de acceso.
Si no recibe la clave de referencia, es posible que haya quedado obsoleto el correo que indicó en su momento. En ese caso escriba a soporteccps@aemps.es para que se lo actualicen. Si lo que está obsoleto es el correo donde se recibe el resto de la documentación de la comunicación (no los localizadores), debe actualizarlo a partir de la modificación “Persona de Contacto: Actualización”. (Punto 11.3.4. del manual de usuario de empresas)
Sí hay posibilidad, si la empresa le autoriza a solicitar sus propias claves para gestionar las comunicaciones. Las claves de usuario se asignan a una persona para una única empresa. Si lleva la asesoría de varias empresas es necesario que nombre a una persona distinta para cada empresa mientras duren sus servicios.
Recuperación de datos de PMPS a CCPS
La aplicación CCPS permite recuperar las comunicaciones existentes en PMPS, actualizando y completando sus datos. La nueva comunicación en CCPS conservará el número que tenía en PMPS. El procedimiento a seguir se explica en el punto 13 del manual de usuario de empresas.
El volcado de comunicaciones de PMPS a CCPS no es automático. Las empresas titulares deben recuperarlas progresivamente a medida que vayan necesitando actualizarlas. La aplicación PMPS se cerró definitivamente el pasado 1 de julio de 2020 por obsolescencia del servidor, por lo que para cumplir con el requisito de mantener las comunicaciones actualizadas será imprescindible la recuperación de cada una de ellas en CCPS.
Las mismas claves que tuviera en PMPS. Las claves de usuario adicionales que se han adjudicado a la misma empresa desde CCPS no son válidas para hacer la recuperación. Una vez recuperada la comunicación en CCPS puede ser gestionada con cualquiera de las claves de usuario de la empresa en CCPS.
Las comunicaciones de PMPS siguen siendo válidas legalmente mientras se encuentren actualizadas, la AEMPS ha determinado que el 4 de julio de 2023., ya han debido recuperar todas las comunicaciones que quedaran en PMPS. Consulten nota informativa al respecto PS, 14/2023
Sí, es posible. Las bases de entidades son comunes para todos los usuarios. Cuando un usuario crea un fabricante que no existía antes, queda a disposición de los demás usuarios que necesiten utilizarlo en sus comunicaciones. Si al pinchar en “Buscar” no aparece ese fabricante, es que tiene que crearlo usted. En ese caso, es importantísimo que introduzca los datos correctamente, incluido el CIF, VAT o equivalente, y que estén completamente actualizados (puede ocurrir que ese fabricante haya cambiado de domicilio o de teléfono desde el último contacto que tuvieron con ellos). Esos datos deben coincidir con los datos de certificado CE, etiquetado e instrucciones de uso. Si hay alguna discrepancia, aclárela con el fabricante antes de introducir sus datos y actualice la documentación si es necesario. Los datos de creación del fabricante nunca deben ser obsoletos. Lo mismo ocurre con el Representante Europeo y con los distribuidores, aunque en el caso de los distribuidores, los datos no aparecen en otros documentos, con lo que basta con que confirme sus datos correctos con ellos.
Si su comunicación está recuperada como borrador, la encontrará en el buzón “Borradores Recuperados”. Solo aparecerá en el buscador general cuando la complete y la envíe a la AEMPS.
Debe completarlas con todas las actualizaciones, tanto de entidades como de otros datos. Es importantísimo que los datos estén actualizados, pues se comparten con todos los usuarios de CCPS. Siempre hay que buscar la entidad (fabricante, representante, distribuidor) antes de crear una nueva, pues es probable que otro usuario la haya creado ya. Hay que revisar todos los datos e introducir las variantes o modelos siguiendo las instrucciones del manual, actualizar el listado de categorías, genéricos y subgenéricos según las nuevas tablas, y completar los datos que no existían en PMPS. Una vez completa y actualizada completamente se envía a la AEMPS. Todo esto se describe con detalle en el punto 13 del manual de usuario de empresas.
No hay otra manera de hacerlo que recuperarlas de PMPS y después darlas de baja. Recupere esas comunicaciones con los datos que faltan y no importa que los certificados estén caducados. MUY IMPORTANTE: si las entidades Fabricante, Representante y Distribuidores no han sido creadas ya por otros usuarios, debe crearlas usted, pero siempre con datos actualizados, pues esos datos son compartidos con todos los usuarios de CCPS y siempre deben estar actualizados. Contacte con las correspondientes entidades antes de crearlas para asegurarse de que los datos son correctos. Una vez su comunicación esté como “Comunicada” ya puede darla de baja. Es importante que lo haga, pues es obligatorio comunicar el cese de la comercialización.
No tiene que pagar ninguna tasa. La recuperación de comunicaciones incluye la posibilidad de actualizar todos los datos necesarios de la comunicación, incluso datos que al modificarse requerirían pago de tasa. Recupere su comunicación con los datos actualizados directamente y no tendrá que pagar ninguna tasa.
El cambio de fabricante de un producto, por definición tanto de la Directiva como del Reglamento, hace que el producto sea otro distinto y que, por tanto, debe comunicarse independientemente del anterior (ver pregunta 7 de la sección ‘Modificaciones’ de esta página). Por tanto, no se puede recuperar de PMPS una comunicación y cambiarle el fabricante. Si ya no comercializa el producto que tenía en PMPS debe recuperarla con los mismos datos y darla de baja. Recuerde que es obligatorio comunicar el cese de la comercialización. Debe hacer una comunicación completamente nueva en CCPS con el nuevo fabricante y pagar la tasa correspondiente.
Comunicaciones en papel anteriores a PMPS
Teniendo en cuenta el tiempo transcurrido desde el 30 de junio de 2013, fecha en que se pudo realizar legalmente la última comunicación de comercialización de productos en papel, que la caducidad de los certificados CE es de cinco años y la obligación de mantener las comunicaciones actualizadas en todo momento, a partir del 28 de junio de 2023 no se podrán ya introducir sin pago de tasa comunicaciones realizadas en papel. Deberán comunicarlas como nuevas respetando la primera fecha de comercialización en España y abonando la tasa correspondiente.
Tasas
Mientras no entre en funcionamiento EUDAMED de forma plena, continúan en vigor los registros nacionales de los productos exactamente en las mismas condiciones que hasta la fecha.
- Registro de Comunicaciones de Comercialización CCPS, destinado a productos sanitarios de las clases IIa, IIb y III (Directiva 93/42/CEE y Reglamento (UE) 2017/745), productos implantables activos (Directiva 90/385/CEE y Reglamento (UE) 2017/745), productos sanitarios para diagnóstico in vitro de las listas A y B y de autodiagnóstico (Directiva 98/79/EC) y clases B, C y D (Reglamento (UE) 2017/746 consolidado) realizadas por cualquier agente económico de cualquier país que comercialice estos productos en España: en estos casos se continuará aplicando UNA TASA POR COMUNICACIÓN y cada comunicación incluye un producto y sus modelos con exactamente la misma consideración de “producto” que en CCPS actualmente; una comunicación puede incluir un UDI-DI o varios. A partir del 28 de junio de 2023 la tasa de epígrafe 8.03 se sustituye por la 5.1, y la tasa 8.33 de modificación desaparece. En lo referente a la aplicación de tramos y la tasa de mantenimiento 5.25 se aplicarán una vez este funcionamiento el nuevo registro de comercialización de productos vinculado a EUDAMED Se mantiene la tasa de nueva comunicación (epígrafe 5.1) para las dos modificaciones individuales que suponen un nuevo producto, según se explica en el Manual de usuario de empresas.
- Registro de Responsables RPS, destinado a productos sanitarios de clase I y productos DIV de cualquier clase, comunicados por sus fabricantes y representantes autorizados establecidos en España, y a las agrupaciones de productos sanitarios realizadas por sus agrupadores establecidos en España. Destinada asimismo a los productos sanitarios a medida por sus fabricantes establecidos tanto en España como fuera de ella y a los representantes autorizados establecidos en España. CONTINÚA EXENTA DE TASAS
Cuando finalmente EUDAMED entre en funcionamiento de forma plena y esté disponible igualmente el nuevo registro de comercialización de productos sanitarios:
- El registro CCPS dejará de estar operativo y será sustituido por el nuevo registro de comercialización de productos sanitarios, aplicable a todas las clases de productos sanitarios y productos sanitarios de diagnóstico in vitro que entren en el ámbito de aplicación de Reglamento (UE) 2017/745 y Reglamento (UE) 2017/746 consolidado y se comercialicen en España. Desde ese momento la tasa inicial 5.1 se aplicará por UDI-DI, en base a las notificaciones por UDI-DI que consten en EUDAMED. Se aplicará el cálculo de tasa por tramos dependiendo del número de comunicaciones (UDI-DIs) que tenga la empresa. También se aplicará la tasa de mantenimiento 5.25 y sus correspondientes tramos de acuerdo a lo indicado en la regulación que establece las tasas de la Agencia. (Ley de garantías y uso racional de los medicamentos y productos sanitarios, aprobado por el Real Decreto Legislativo 1/2015, de 24 de julio, modificado por la Ley 38/2022, de 27 de diciembre
- El Registro RPS quedará reservado únicamente a productos sanitarios a medida y continuará exento de tasas. El resto de productos se comunicarán en el nuevo Registro de Comercialización abonando la tasa correspondiente.
Todas las dudas relacionadas con el pago de las tasas pueden resolverse consultando en la dirección tasas. Los sistemas de pago son muy variados y están descritos en dicha página.
Se recomienda el pago telemático cuya validación es automática y permite el envío inmediato de la comunicación. El pago se realiza exclusivamente desde la propia aplicación CCPS pinchando el enlace “Pague aquí su tasa”
Hay que abonar una tasa para cada comunicación de comercialización y cada comunicación solo puede incluir un producto sanitario (aunque, en ocasiones, un producto puede incluir varias variantes o modelos del mismo producto, pero nunca varios productos distintos, salvo que éstos se comercialicen y utilicen conjuntamente como un sistema o como una agrupación de productos). Puede consultar estos aspectos en el manual de usuario en los puntos 5.2.1.3 y 5.2.1.4.
Desde el 25 de septiembre 2023, el único motivo de exención de tasa se aplica a las comunicaciones de componentes de una agrupación que no se comercializan por separado, ver punto 7.1. del Manual de usuario de empresas. Estas comunicaciones no emiten justificante de comunicación al ser únicamente una herramienta para comunicar la agrupación que sí paga tasa y sí emite justificante.
Desde la entrada en funcionamiento de la versión 2.0.8 de la aplicación, el 28 de junio 2023, la exención de tasas se detecta automáticamente por la aplicación. La casilla “Exenta de tasas”, aparecerá marcada cuando el sistema detecta que puede acogerse a motivo de exención.
Desde el día 28 de junio 2023 con motivo de la entrada en vigor de la nueva Ley 38/2023 .de tasas., ha cambiado la gestión de pago de tasas para que se realice desde las propias aplicaciones.
Debe acceder a la aplicación CCPS, abrir el borrador y, solo cuando ya lo haya completado, pinchar el enlace “Pague aquí su tasa” en la pestaña de “Datos Generales”.
Al gestionar la tasa y volver a la aplicación, aparecerán automáticamente los datos de la tasa generada, no tiene que escribir ningún dato ni adjuntar ningún justificante.
Puede consultar la última edición del Manual de usuario de empresas. que lo explica con detalle y elManual Sede (aemps.gob.es) del Servicio de Tasas de la AEMPS.
El sistema de pago ha cambiado desde el pasado 28 de junio 2023, se describe con detalle en el manual de usuario CCPS última edición colgado en la aplicación.
Si la tasa aparece como “Pendiente” no tiene que hacer nada, solo esperar a que Tasas valide el documento. Cuando eso ocurra su comunicación pasará al buzón “Borradores con tasa pagada” En ese momento podrá ya enviar el borrador a la AEMPS.
Para facilitar el proceso se recomienda el pago telemático con el que la validación es automática y el borrador pasa inmediatamente a “Borrador con tasa pagada pendiente de envío”.
Elaboración de la comunicación
Los productos sanitarios de clase I, y sus variantes (Is, Im, Is+m, Ir), así como los productos sanitarios de diagnóstico in vitro de autocertificación por el fabricante (grupo “otros” de la Directiva 98/79 o clase A y A estéril del Reglamento DIV), no se comunican en CCPS. Es por esa razón por la que no lo encuentra. Solo debe comunicarlo en la aplicación RPS, en caso de que usted sea su fabricante o su representante autorizado, y esté establecido en España. Si no es el caso no tiene que comunicarlos en la AEMPS. Puede consultar la información sobre RPS en Introducción en el mercado europeo (RPS).
No, nunca debe crear un fabricante que ya existe en la base con otros datos. Compruebe si los datos que usted tiene están actualizados o están obsoletos. Si los datos son obsoletos, póngase en contacto con el fabricante para que le envíe documentación actualizada (certificado CE, etiquetas e IFU). Si sus datos están actualizados y los que hay en la aplicación son antiguos, póngase en contacto con soporteccps@aemps.es.
En caso de que por error haya introducido un importador y desee borrarlo, pulse “Cambiar” y, en el buscador, realice una búsqueda cualquiera para que aparezca el botón de crear importador. Cuando aparezca la ventana para introducir los datos del importador nuevo, pulse “Cancelar” y se le borrará el importador que tuviese seleccionado. Esto solo se puede realizar en el estado “Borrador de la comunicación”. Una vez enviada, si se ha confundido de importador debe sustituirlo por el correcto, aunque no sea español, utilizando la modificación “Importador Sustitución”.
-
En los datos de la empresa comunicante figura el CIF de la empresa XXX y no puede introducirse el de la empresa YYY.
Las claves de acceso, a todos los efectos, son propiedad y responsabilidad de la empresa comunicante, en este caso XXX, por eso aparece siempre su CIF. La empresa, bajo su exclusiva responsabilidad, puede solicitar más claves de usuario para su misma cuenta y tener así varios gestores.
-
En los datos del comunicante “en calidad de”, ¿qué hay que indicar si se trabaja como un asesor externo?
En los datos del comunicante, “en calidad de”, se refiere siempre a la empresa comunicante, nunca a la persona que hace la comunicación en su nombre. La relación que se pide es la de la empresa comunicante (XXX) respecto del producto que está comunicando, por lo que solo puede ser fabricante del producto, representante autorizado del fabricante en UE, importador del producto, distribuidor del producto o agrupador.
En la medida de lo posible, debe seleccionar las opciones de los desplegables que se encuentran en la pestaña de productos y solo en caso necesario, seleccionar “Otros”. En caso de seleccionarla, deberá especificar una característica que defina ese campo de la forma más breve posible, y que defina la característica más notoria del mismo. Ej: Genérico: Hemodializador; Tipo: Otro; Otro especificar: de fibra.
El tiempo máximo de inactividad es de 60 minutos, por lo que se recomienda guardar a menudo los datos introducidos para evitar pérdida de información y empleando el botón “Guardar”. De este modo, se conservan los datos introducidos en la comunicación. Asimismo, la información se mostrará en las distintas pestañas de la comunicación en su fase de borrador. La comunicación guardada aparecerá en el buzón de borradores y se guardan pestaña por pestaña. Debe completar los datos de la pestaña entera para poder guardar el borrador.
Informe de dicha incidencia en la página https://servicedesk.aemps.es/servicedesk/customer/portal/2/ incluyendo una copia de la pantalla de error. Para ello, pulse el botón del teclado “Impr Pant” que suele estar a la derecha del teclado junto a la línea de teclas F (F1, F2, etc.).
Los accesorios, en el caso de ser un producto incluido dentro del producto sanitario, deben incluirse en el apartado “Productos incluidos”. Por ejemplo: en un PSIA, un software sería un producto incluido. En el caso de tratarse de un producto sanitario diferente del que está comunicando y que se comercializa por separado, deberá hacer una nueva comunicación de comercialización en caso de ser clase IIa, IIb o III.
La adjudicación de categorías se realiza por orden, incluyendo el producto en el primer grupo en el que puede entrar, así, por ejemplo, aunque los electrodos de ablación son productos de un solo uso, también son productos electromédicos mecánicos porque utilizan electricidad y esta categoría, nº 4, aparece antes que la nº 10 de un solo uso. Por tanto, es correcto que esté en el grupo 4.
Algunos productos de clase IIa son muy sencillos y no requieren folleto de instrucciones de uso separadas. No obstante, por sencillo que sea siempre hay que especificar unas precauciones mínimas de utilización y/o advertencias. Estas suelen ir impresas en el envase. En estos casos, deben incluir la copia de la etiqueta del envase donde aparezcan estas advertencias como instrucciones de uso. La aplicación exige que siempre se incluyan estas instrucciones y deberá introducirlas, aunque sean una copia del etiquetado.
Agrupaciones
Una agrupación es un conjunto de varios productos sanitarios, de uno o de distintos fabricantes, que ostentan cada uno de ellos su marcado CE (han sido certificados independientemente unos de otros, no como kit), empaquetados conjuntamente y que se ponen en el mercado por un tercero (agrupador) como sistemas, conjuntos o equipos para procedimientos médicos o quirúrgicos. La finalidad de uso del conjunto debe ser la de Producto Sanitario. En las agrupaciones acogidas al Reglamento (UE)/2017/745) pueden intervenir componentes tanto de la Directiva de PS como del Reglamento de PS y productos del Reglamento de DIV. Este concepto no es aplicable a los kits cuya finalidad de uso es de DIV. En las agrupaciones de Directiva solo se pueden incluir componentes de Directiva. (Ver pregunta de este epígrafe). Este concepto no es aplicable a los productos sanitarios de diagnóstico in vitro; no existen las agrupaciones DIV, aunque un DIV de reglamento sí puede formar parte de una agrupación de PS de reglamento cuando la finalidad médica del conjunto es de producto sanitario.
En CCPS comunica cualquier agente económico que comercialice en España una agrupación que contenga componentes de clase IIa, IIb o III, según el artículo 22 del RD 1591/2009 y, con la entrada en vigor de los reglamentos, algún componente DIV si cumple el reglamento DIV.
En RPS solo comunican los agrupadores establecidos en España (tienen obligación de tener licencia de IPS para poder realizar su actividad de agrupación). Comunican todas las agrupaciones que ellos realicen, independientemente de si se comercializan en España o no y de la clasificación de los componentes de cada agrupación, en base al artículo 24 del RD 1591/2009.
Puede darse el caso de que una empresa tenga que comunicar la misma agrupación en las dos aplicaciones si cumple los requisitos de los dos tipos de comunicación, es decir, el caso de los agrupadores establecidos en España que comercialicen en España una agrupación de las que requiere comunicación en CCPS.
Debe realizar una comunicación independiente de cada uno de los componentes de la agrupación. Si esos componentes no se comercializan por separado, esas comunicaciones están exentas de tasa y no generan documentación de comunicación, solo son un instrumento para poder realizar la comunicación de la agrupación. La tasa se abonará por la comunicación de la agrupación que incluye a las individuales. Si, por el contrario, ese componente se comercializa también independientemente, pagará su tasa y será una comunicación normal que generará toda la documentación correspondiente a cualquier comunicación. Vea con detalle el punto 7 del manual de usuario de la aplicación donde se explica.
Si la finalidad de uso del conjunto es la de diagnóstico in vitro, el fabricante del DIV es responsable del conjunto DIV+PS, según el punto 1.6 del “IVD Medical Device Borderline and Classification issues, MEDDEV 2.14/1 revision 2 January 2012”. No se aplica el concepto de “Agrupación” y cada producto tiene que cumplir su propia regulación y debe ser comunicado en CCPS si el tipo de producto lo requiere (DIV lista A, lista B o autodiagnóstico si son de Directiva, y clase B, C o D si son de Reglamento y PS IIa, IIb o III). Estos productos se comunican por separado, cada uno en su apartado correspondiente, y se incluye una referencia cruzada al número de comunicación que tiene el otro componente en el apartado “Observaciones” de cada comunicación. Todo esto se explica con detalle en el punto 8 del manual de usuario CCPS.
Si la finalidad de uso del conjunto es la de producto sanitario, y el producto sanitario de diagnóstico in vitro cumple el Reglamento (UE) 2017/746, sí que puede agruparse con un producto sanitario en un pack de procedimientos del artículo 22 del Reglamento (UE) 2017/745. Se comunican como “Agrupación” siguiendo las instrucciones del punto 7 del manual de usuario CCPS.
A partir del 26 de mayo 2021 con la entrada en vigor del Reglamento (UE) 2017/745 de Productos Sanitarios, las agrupaciones deben cumplir ya el artículo 22 del Reglamento, pero pueden incluir tanto productos certificados con arreglo a los Reglamentos de PS y DIV como productos de Directiva 93/42/CEE que puedan acogerse al artículo 120 del Reglamento (UE) 2017/745. Por otra parte, de acuerdo al documento MDCG 2021/25, si todos los productos que componen la agrupación se acogen al artículo 120 del Reglamento mencionado, la agrupación completa puede continuar acogida al artículo 12 de la Directiva 93/82/CEE en lugar del art. 22 del Reglamento (UE) 2017/745.
Puede consultar los detalles en el punto 8 del manual de usuario CCPS.
Es necesario tener en cuenta el procedimiento previsto para ellas. Se podrán recuperar parte de los datos y documentación, pero habrá que crear las comunicaciones de los componentes. El orden a seguir es el siguiente:
Primero, crear nuevas comunicaciones completas de los componentes (estos no existían como tales en PMPS, salvo si se comercializaban también individualmente, en cuyo caso sí se recuperarían normalmente), y enviarlas a la AEMPS. No olvide marcar la casilla “Componente de agrupación” en la pestaña “Datos Generales”. Las comunicaciones aparecerán como “Huérfanas” hasta que queden asociadas a la agrupación.
Después, recuperar la comunicación de PMPS de la agrupación, en la que deberá eliminar la documentación de certificados CE de los componentes y etiquetados de los mismos (puesto que estos formarán parte de las comunicaciones de los componentes, ya realizadas en el punto anterior), así como los datos de fabricante y representante europeo si procede. Esta será la comunicación que tendrá el número de PMPS recuperado. En esta comunicación recuperada marcará la casilla “Agrupación” y asociará las comunicaciones de los componentes creadas y enviadas anteriormente. Una vez completa se envía a la AEMPS. En ese momento, las agrupaciones de los componentes anteriores quedan vinculadas a la agrupación y desaparecerán del buzón de “Huérfanas”.
El procedimiento de recuperación se describe con detalle en el punto 13 del manual de usuario de empresas. Teniendo en cuenta que el 3 de julio de 2018 la aplicación PMPS dejó de estar operativa, la obligación de mantener actualizadas las comunicaciones y que los certificados CE tienen un periodo máximo de validez de cinco años, el 4 de julio de 2023 no debe haber más comunicaciones en PMPS sin recuperar.
En este caso no puede utilizar la comunicación ya realizada como componente para la agrupación, puesto que incluye modelos que no van en la agrupación. Debe realizar una comunicación exclusivamente con el modelo de producto que entra como componente de la agrupación, marcando “No se comercializa independientemente” para no pagar tasa de nuevo, puesto que ese producto ya tiene su propia comunicación con tasas.
Algunos datos de la agrupación se toman directamente de los componentes que lo forman, como son los relativos a los certificados CE, los fabricantes y los representantes europeos autorizados. Por tanto, para modificar uno de esos datos de la agrupación se debe hacer la modificación en la comunicación del componente que esté afectado, y los datos del componente modificados se volcarán directamente en la comunicación de la agrupación. En el manual de usuario de la aplicación se indica cuáles son las modificaciones concretas a las que se refiere este punto. Solo hay una modificación que no es posible hacerla en agrupaciones: “Cambio de comunicante (traspaso de comunicaciones)”. Esas comunicaciones deben darse de baja por el comunicante que ya no las va a comercializar y debe hacerlas nuevas el nuevo comunicante.
Para que la aplicación pueda asociar la agrupación tiene que haber sido marcada previamente como componente de agrupación. Si no lo fue porque se creó con anterioridad, escriba a soporteccps@aemps.es indicando el número de comunicación que hay que marcar para que lo seleccionen desde la AEMPS.
En un componente de agrupación deben indicar en calidad de Distribuidor, Importador Fabricante o Representante, según proceda, nunca como Agrupador. Esta cualidad solo es aplicable a la comunicación de la agrupación completa, no a sus componentes.
Particularidades de las comunicaciones de productos sanitarios DIV
El analito es el objeto del test. Ej.: Chlamydia, glucosa, etc.
En la mayoría de los casos, los productos de diagnóstico in vitro constan de dos fases principales:
- Desarrollo/ metodología de la técnica:
- Que podría ser serológico (si la diana a la que va dirigido el test son antígenos o anticuerpos) o,
- Por hibridación/amplificación (PCR, entre otras): ADN, ARN, cromosomas, etc. Es decir, por amplificación de ácidos nucleicos (DNA, RNA…) o por hibridación génica a una sonda marcada que no conlleva amplificación.
- Detección de la técnica: según el tipo de marcador usado para dar el resultado como CLIA (si el marcador es quimioluminiscente), ELISA (si es una enzima), RIA (si es un radioisótopo…).
Para agrupar mejor estos productos, se han establecido categorías duales. Ejemplo: si su producto es para detectar el antígeno p24 del VIH y la detección es por quimioluminiscencia, deberá seleccionar: “Serológico (detección de Ag o Ac) + CLIA (Quimioluminiscencia)”.
Existen otros casos en los que no es necesario determinar el desarrollo previo por la especificidad del propio método, por lo que no tienen representación dual, por ejemplo: inmutinción, inmunocromatografía, etc.
En estos casos puntuales, deberá seleccionar como “Tipo de muestra: No Procede (opción incluida dentro del desplegable), Método Analítico: Software”. El tipo de resultado dependerá de si es numérico (cuantitativo) o cualitativo. El campo “Analito” debe cumplimentarse según la relación de productos obligados a comunicarse; los que están dentro de lista A (como VIH, por ejemplo), lista B (como trisomía del par 21) o autodiagnóstico. Le servirá de ayuda el certificado CE expedido por el organismo notificado que lo habrá clasificado en la lista correspondiente o como autodiagnóstico.
Tal y como viene indicado en el RD 1662/2000 relativo a los productos para diagnóstico in vitro (anexo II), este tipo de productos están incluidos en la lista B. Por tanto, deberá seleccionar: “Clase: Lista B; Genérico: Glucemia” (para Autodiagnóstico).
Los test de VIH pertenecen siempre a la lista A, aunque sean de autodiagnóstico. Prevalece siempre el riesgo de la lista A, por eso podrá encontrarlos en el desplegable de lista A, no en la de autodiagnóstico. Así lo verá también en el certificado CE expedido por el organismo notificado.
La mayoría de los tests de embarazo se basan en una inmunocromatografía, en que el analito a detectar es la hCG (gonadotropina coriónica humana), por tanto, deberá seleccionar las siguientes opciones:
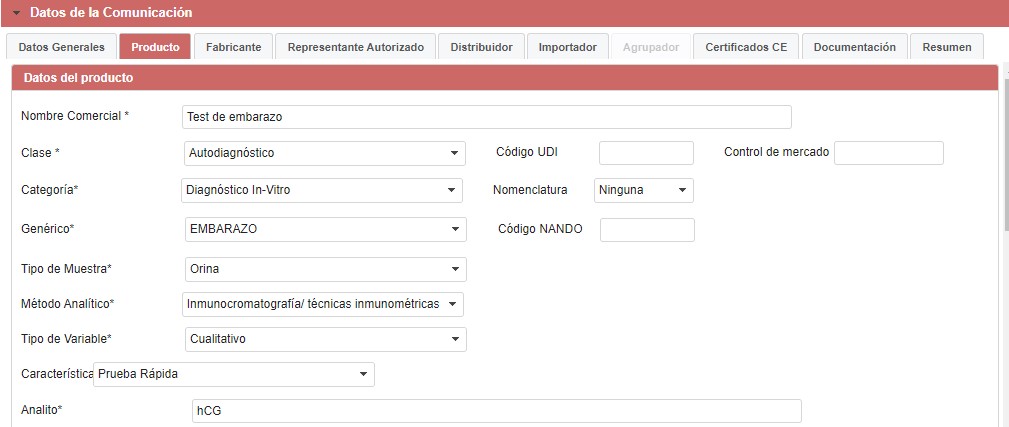
En “Productos incluidos” deben detallar todos los componentes que se incluyen en el envase pero no están premontados, por ejemplo: varilla, cuentagotas, pipeta o lo que lleve su producto, además del producto principal.
Si su producto no tiene alguna de las siguientes características, debe seleccionar “Otros” e indicar “No procede”:
- Carga vírica
- Prueba rápida
- Test genético
- Calibración
- Control
Modificaciones
La gestión de modificaciones se ha pensado con el objetivo de facilitar al máximo la actualización de sus comunicaciones. Es por ello que:
- sólo dos llevan tasa: las modificaciones individuales que implican en realidad un producto distinto: “Productos incluidos: ampliación/eliminación” y “Componente de agrupación que pasa a comercialización separada”.
- existen las modificaciones múltiples. Ninguna paga tasa.
- Incorporan automáticamente los nuevos datos de la modificación y la nueva documentación en la comunicación al terminar el proceso. Para ello, asegúrese siempre de que la modificación ya no se encuentra en la bandeja “Pendiente actualizar documentación”.
Existen dos tipos, individuales y múltiples.
- Las modificaciones individuales corresponden a aquellas que solo afectan a un producto en concreto.
- Las modificaciones múltiples suponen una modificación que afecta o podría afectar a más de un producto.
Por ejemplo, la ampliación de variantes afecta a un solo producto, mientras que, la actualización del domicilio de un representante autorizado afecta a todas las comunicaciones en las que figure dicha empresa.
Las CCPS afectadas por las modificaciones múltiples que conllevan inclusión de documentación (afectan a fabricante, representante autorizado, organismo notificado o revalidación de certificado CE) pasarán al buzón “Pendiente de actualizar documentación”, desde el cual usted podrá incluir en cada una de ellas la documentación (etiquetado, instrucciones de uso, certificados) con esa nueva información, a medida que vaya disponiendo de ella. La modificación de datos se realizará en todas las CCPS en las que esos datos estén incluidos.
Para realizar este cambio debe seleccionar en el menú de la izquierda el apartado “Modificaciones” y, una vez allí, seleccionar en “Modificaciones múltiples sin tasa”, “Persona de contacto: actualización”. Allí podrá modificar todos los datos correspondientes a la persona que realiza la comunicación. Para realizar esta modificación, como cualquier otra, asegúrese de que la dirección de correo de seguridad de las claves de usuario está actualizada. Si no recibe la clave de referencia, póngase en contacto con soporteccps@aemps.es. Puede consultar un ejemplo en el manual de usuario de la aplicación (punto 11.3.4).
Esta posibilidad la puede hacer como una modificación individual. Para ello, deberá seleccionar “Modificaciones”, y en la lista de modificaciones individuales, seleccionar “Nombre comercial: Sustitución”. Esta modificación no paga tasa y requiere actualización de etiquetas e instrucciones de uso. Al finalizar la tramitación recibirá nuevo justificante de comunicación con el nuevo nombre.
Si la empresa fabricante ha cambiado de domicilio de fabricación, pero está dentro de la misma compañía, es decir el fabricante legal sigue siendo el mismo y en el certificado CE sigue apareciendo el mismo domicilio social, se trataría de un cambio de planta de fabricación dentro de la misma empresa.
La planta de fabricación no es un dato de la comunicación, solo lo es el domicilio del fabricante legal, titular del certificado CE. Tendría que actualizar el certificado CE si la planta nueva no aparecía en él (“Certificado CE actualización por revalidación“). En cuanto al etiquetado e instrucciones, solo tendría que actualizarlos si la planta de fabricación aparece. Este es un dato voluntario del etiquetado e instrucciones.
Si por el contrario lo que cambia es el domicilio social del fabricante legal, titular del certificado CE, tendrá que hacer una modificación múltiple de “Fabricante: actualización de domicilio y/o nombre“. Esta modificación se aplica a todas las comunicaciones que tenga ese fabricante y no es posible seleccionar unas sí y otras no, puesto que se trata siempre del domicilio del mismo fabricante legal.
La actualización de domicilio y/o nombre se refiere a cambios de emplazamiento de la misma empresa fabricante o a cambios en su denominación (por ejemplo, EMPRESA S.L. cambia a EMPRESA S.A.), pero siempre y cuando se trate de la misma empresa titular del certificado CE. Requerirá la emisión de un anexo al certificado CE indicando esta circunstancia, o bien un nuevo certificado con los nuevos datos. Todos los productos de esa empresa cambian a los nuevos datos, no hay posibilidad de selección puesto que es la empresa entera la que cambia sus datos y, por tanto, todas sus comunicaciones. Se incluyen en este caso las absorciones completas de una empresa fabricante por otra.
El cese de distribución se formaliza en las comunicaciones como solicitud de baja. Busque su comunicación con el buscador general del menú de la izquierda y entre en su comunicación. En la parte inferior de la pantalla verá un botón “Baja”. Si lo pulsa, el sistema pedirá el motivo de la baja, se rellena, se pulsa “Aceptar” y la empresa recibirá un oficio de baja. La comunicación pasará al estado “Baja”. Las bajas motivadas por decisión de la empresa debe gestionarlas directamente la empresa, no la AEMPS. Si la comunicación que desea dar de baja está en PMPS y no ha sido recuperada aún, debe recuperarla primero para darla de baja desde CCPS. Se recuerda la obligación de comunicar el cese de la comercialización. Las únicas bajas que no puede dar la empresa son las de las agrupaciones y los componentes de agrupación. Estas debe solicitarlas al buzón soporteccps@aemps.es indicando el número de comunicación y el motivo de la baja.
Hasta que la empresa receptora del traspaso no comercialice los productos recibidos de la primera empresa, no puede hacer la comunicación en la aplicación. La fecha que debe registrar la aplicación nunca puede ser anterior a la de comercialización real de los productos por parte de la empresa receptora. La obligación de comunicar la comercialización de un producto siempre es posterior a la comercialización, nunca anterior.
La obligación de comunicar la comercialización de un producto siempre es posterior a la comercialización, nunca anterior. De este modo, nunca va a haber necesidad de cambiar la fecha de comercialización por error o cambio de planes de comercialización de la empresa. Si hay que cambiar la fecha de comercialización, se considera una nueva comunicación y conlleva dar de baja la comunicación anterior y pagar la tasa de nueva comunicación.
Se refiere a cualquier cambio de los productos que componen la comunicación, por ejemplo, los productos incluidos en una agrupación o los componentes que acompañan al producto sanitario comunicado, sin los cuales no puede funcionar. Estos cambios pueden ser de cualquier tipo, en sus características o en su presencia o no en la comunicación. Esta modificación genera un producto distinto del inicial, por lo que su modificación implica el abono de una tasa de modificación. Seleccione la modificación individual “Productos incluidos: Ampliación/Eliminación”. Se describe en el punto 11.2.3. del manual de usuario de la comunicación.
La composición cualitativa de un reactivo es crítica para definir de qué producto se trata, por tanto, si la composición cualitativa varía, se considera un nuevo producto. Para tramitarlo debe dar de baja el producto anotado y realizar una nueva comunicación abonando la tasa correspondiente a nueva comunicación.
Seleccione la modificación “Certificado CE: Actualización por reclasificación del producto.” Se explica con detalle en el punto 11.2.1. del manual de usuario de la aplicación.
Aunque una comunicación se haga en calidad de distribuidor, no se incluyen automáticamente los datos de esa empresa comunicante en la pestaña correspondiente. Debe incluirse expresamente en la pestaña “Distribuidores”. Una vez comunicada, puede hacerlo sin ningún coste utilizando el menú “Modificaciones” y seleccionando la modificación múltiple “Distribuidor: ampliación/eliminación”, siguiendo las instrucciones del manual de usuario de la aplicación (punto 11.3.9).
Lo mismo ocurre si realiza la comunicación en calidad de otra figura, por ejemplo “Fabricante”. En cualquier caso, deberá rellenar los datos de la pestaña correspondiente (“Fabricante”), pues su cumplimentación no es automática.
Actualización certificados CE
Puede realizar directamente la modificación múltiple “Certificado CE: actualización por revalidación” seleccionando la opción “Caducidad del certificado” si lo único que ha cambiado es la fecha de validez o el número de certificado.
La gestión de la comunicación debe hacerse utilizando el botón “Modificaciones” y seleccionando la modificación correspondiente. Hay varias posibilidades:
- Modificación múltiple (aunque solo la realice en una comunicación) “Actualización certificado CE por revalidación”. Es la modificación más habitual; simplemente ha caducado el certificado o, aunque no haya caducado, se ha emitido otro que lo sustituye en el que no cambia ninguno de los datos oficiales: mismo ON, mismos datos de fabricante y mismos datos de representante cuando lo incluye, mismo procedimiento de certificación y misma clasificación del producto. Se explica en el punto 11.3.1. del manual.
- Modificación individual: “Actualización certificado CE por reclasificación”. Solo se utiliza cuando el producto ha cambiado de clasificación, por ejemplo, si pasa de clase IIa a clase IIb. Se explica en el punto 11.2.1. del manual. (Muy poco frecuente).
- Modificación individual: “Actualización del certificado CE por cambio en el procedimiento de certificación”. Solo se utiliza cuando el fabricante cambia de ruta de certificación, por ejemplo, de Anexo V+ Declaración CE de conformidad pasa a Anexo II punto 3. Se explica en el punto 11.2.2. del manual. Tampoco es muy frecuente, aunque algo más que el caso anterior.
- Actualizaciones del certificado CE que incluyan además cambios en datos oficiales, por ejemplo, domicilio del fabricante legal o número de ON que lo certifica, o datos del representante, o sustitución de un representante por otro. En todos esos casos debe seleccionar las modificaciones correspondientes, que son prioritarias respecto a las del certificado. Todas ellas incluyen la necesidad de actualizar el certificado CE, etiquetas e instrucciones de uso con los nuevos datos. Son modificaciones múltiples y la actualización de documentos se hace una por una en cada comunicación.
- Actualización del certificado CE por cambio de legislación, de directiva a reglamento. Se llama “Cambio de legislación a reglamento” y es una modificación individual que se explica en el punto 11.2.6. del manual de usuario CCPS.
Desde la entrada en vigor del Reglamento (UE) 2023/607 el 20 de marzo 2023, ya no se emiten nuevos documentos de artículo 97 por las autoridades sanitarias. Ese documento era uno de los requisitos opcionales para los certificados que caducaron antes del 20 de marzo 2023.
Si el fabricante opta por no acogerse a la extensión de validez del Certificado CE de acuerdo con el Reglamento (UE) 2023/607 y el Reglamento (UE) 2024/1860, y no va a continuar comercializando el producto una vez finalizado el stock, seleccione la modificación “Certificado CE por revalidación” y seleccione la opción “Stock de producto fabricado antes de caducidad de certificado CE”. Los datos del certificado CE deberán mantenerse, sin modificarlos. En la fase de actualización de la documentación, además del documento del certificado CE debe añadirse un documento adicional donde indique cuántos lotes y unidades de cada uno quedan en stock en sus almacenes amparados por ese certificado, y un texto que explique que todos esos lotes han sido fabricados antes de la fecha de caducidad del certificado CE.
Cuando finalice el stock de sus almacenes debe darse de baja la comunicación.
Si los productos reúnen los requisitos establecidos por el Reglamento (UE) 2023/607 y el Reglamento (UE) 2024/1860 para extender la validez de su certificado CE de acuerdo con su clasificación según lo establecido en dichos reglamentos, siga estas instrucciones:
- Seleccione la modificación Certificado CE por revalidación, marcando la opción “Los productos se acogen a extensión de validez de certificado CE” y manteniendo los datos del certificado sin modificar.
- En la fase de actualización de documentación, además del certificado CE deben añadirse los documentos que avalan que se cumple los requisitos del correspondiente Reglamento:
- El acuerdo escrito de conformidad entre el ON y el fabricante, firmado en los plazos recogidos en el Reglamento que sea de aplicación.
- Declaración del fabricante, emitida bajo su exclusiva responsabilidad, en la que confirma que cumple con las condiciones para acogerse a los periodos transitorios que apliquen, indicando la fecha de finalización del periodo de transición e identificando los productos cubiertos y los certificados correspondientes. La declaración incluirá lo siguiente:
- Que los productos siguen cumpliendo lo dispuesto en la Directiva 93/42/CEE, Directiva 90/385/CEE o Directiva 98/79/CE según proceda.
- No hay cambios significativos en el diseño ni en la finalidad prevista de los productos desde el 26 de mayo 2021 en el caso de productos sanitarios y desde el 26 de mayo de 2022 para productos sanitarios para diagnóstico in vitro.
- Los productos no presentan un riesgo inaceptable para la salud o la seguridad de los pacientes, usuarios u otras personas o para otros aspectos de la protección de la salud pública.
- El fabricante dispone de un sistema de calidad de acuerdo al artículo 10 (9) del MDR a más tardar desde el 26 de mayo de 2024 en el caso de productos sanitarios o de acuerdo al artículo 10 (8) del IVDR a más tardar desde el 26 de mayo 2025 en el caso de productos sanitarios para diagnóstico in vitro.
Cuando obtenga se obtenga el certificado CE de acuerdo con Reglamento, haga entonces la modificación normal de Cambio de legislación a Reglamento como se explica en el Manual de usuario de empresas.
No cualquier certificado CE caducado es válido. Tiene que cumplir una serie de requisitos que se establecen en el Reglamento (UE) 2023/607 y en el Reglamento (UE) 2024/1860.
En el caso de certificados caducados que se acogen a la extensión de validez de acuerdo con el Reglamento (UE) 2023/607, estos serán los siguientes:
- Haber sido emitido en base a la Directiva 93/42/CEE después del 25 de mayo de 2017.
- Continuar en vigor el 26 de mayo 2021 y no haber sido retirado.
- Cumplir todas las condiciones para poder acogerse al artículo 120 del Reglamento (UE) 2017/745.
- Si el certificado CE fue emitido entre el 25 de mayo 2017 y el 25 de mayo de 2021 y, dependiendo de si caducó antes o después del 20 de marzo de 2023, deben cumplir requisitos específicos y que se indican en el Reglamento (UE) 2023/607.
En el caso de certificados CE caducados que se acogen a la extensión de validez de acuerdo con el Reglamento (UE) 2024/1860, estos serán los siguientes:
- Haber sido emitido en base a la Directiva 98/79/CE después del 25 de mayo de 2017.
- Continuar en vigor el 26 de mayo de 2022 y no haber sido retirado.
- Cumplir todas las condiciones para poder acogerse al artículo 110 del Reglamento (UE) 2017/746.
- Si el certificado CE fue emitido entre el 25 de mayo de 2017 y el 25 de mayo de 2022 y, dependiendo de si caducó antes o después del 9 de julio de 2024, deben cumplir requisitos específicos y que se indican en el Reglamento (UE) 2024/1860.
En el caso de los productos sanitarios, los periodos transitorios de validez de los certificados CE emitidos de acuerdo con Directiva se recogen en el Reglamento (UE) 2023/607 del Parlamento Europeo y del Consejo de 15 de marzo 2023. De acuerdo con este Reglamento, se extienden los periodos transitorios y la validez de los certificados CE en base a la clase de riesgo de los productos acogidos al Reglamento (UE) 2017/745 (MDR) bajo determinadas condiciones del siguiente modo:
- Hasta el 31 de diciembre 2027 para los productos de clase III y IIb Implantable (excepto material de sutura, grapas, material para obturación dental, aparatos de ortodoncia, coronas dentales, tornillos, cuñas, placas, cables, alfileres, clips y dispositivos de conexión).
- Hasta el 31 de diciembre 2028 para el resto de los productos.
- Hasta el 26 de mayo 2024 para los productos que no se van a acoger al Reglamento después de esa fecha.
En el caso de los productos sanitarios para diagnóstico in vitro, los periodos transitorios de validez de los certificados CE emitidos de acuerdo con Directiva se recogen en el Reglamento (UE) 2024/1860 del Parlamento Europeo y del Consejo de 13 de junio de 2024. De acuerdo con este Reglamento, se extienden los periodos transitorios y la validez de los certificados CE en base a la clase de riesgo de los productos acogidos al Reglamento (UE) 2017/746 (IVDR) bajo determinadas condiciones del siguiente modo:
- Hasta el 31 de diciembre de 2027 para los productos que tengan un certificado CE que haya sido expedido conforme con la Directiva 98/79/CE y sea válido de acuerdo con el artículo 110.2 del Reglamento (UE) 2017/746 modificado por el Reglamento (UE) 2024/1860.
- Los productos cuyo procedimiento de evaluación de la conformidad con arreglo a la Directiva 98/79/CE no haya requerido la participación de un organismo notificado, para los que se haya elaborado una declaración de conformidad antes del 26 de mayo de 2022 con arreglo a dicha Directiva, y para los cuales el procedimiento de evaluación de la conformidad con arreglo al Reglamento 2017/746 requiera la participación de un organismo notificado se podrán introducir en el mercado o poner en servicio:
- Hasta el 31 de diciembre de 2027 en el caso de los productos de la clase D
- Hasta el 31 de diciembre de 2028 en el caso de los productos de la clase C
- Hasta el 31 de diciembre de 2029 en el caso de los productos de la clase B y en el caso de los productos de la clase A introducidos en el mercado en condiciones estériles.
Entre en la comunicación y compruebe que ha eliminado el certificado caducado, o en caso de extensión de validez de este, o de existencia de stock fabricado cuando este estaba en vigor, que se ha marcado la casilla correspondiente (“Los productos se acogen a extensión de validez de certificado CE” o “Stock de producto fabricado antes de caducidad ce certificado CE”) manteniendo el certificado.
De lo contrario, seguirá apareciendo el aviso “Certificados caducados” en el buzón de la pantalla inicial. Si es así repita la modificación “Certificado CE actualización por revalidación” y elimine el documento caducado, o marque la casilla correspondiente. En el histórico de la comunicación se pueden consultar los documentos no vigentes si fuera necesario.
Dudas sobre la aplicación de la legislación
Solo se comunican en CCPS los productos que tienen ya un certificado CE de la Directiva 93/42/CEE. Si el certificado caduca y no se renueva, hay que seguir las instrucciones previstas para los productos en esta situación.
Los productos sin finalidad médica que no tenían certificado CE de directiva disponen de un plazo para obtener el certificado CE emitido por un organismo notificado, con arreglo al Reglamento hasta el 31 de diciembre 2028 si no requieren la realización de ensayos clínicos, y hasta el 31 diciembre 2029 si necesitaran dichos ensayos siempre y cuando hayan iniciado sus Investigaciones clínicas antes del 31 diciembre 2027. Los productos no se comunicarán hasta disponer de este certificado. (Para más información, ver la nota informativa PS, 38/2022 publicada en la web de la AEMPS y el Artículo 2 del Reglamento de Ejecución (UE) 2022/2346 de la Comisión de 1 de diciembre de 2022 para estos productos, modificado por el Reglamento de Ejecución (UE) 2023/1194 de la Comisión de 20 de junio de 2023)
La puesta en el mercado se refiere a la primera comercialización de un producto que se realiza en España, con lo cual con comunicar una vez ese producto por cualquiera de los agentes económicos implicados (fabricante, distribuidor, importador, agrupador) era suficiente. A partir del 21 de marzo de 2010, con la entrada en vigor de los nuevos reales decretos, la obligación cambia a comunicar la comercialización de los productos IIa, IIb y III. La comunicación de comercialización consiste en que cada uno de los agentes económicos tiene que comunicar la primera comercialización de un producto en España. Es decir, con la legislación anterior, con que un producto estuviera comunicado por algún agente era suficiente, pero a partir del 21 de marzo de 2010, todos los agentes que comercialicen un producto en España están obligados a comunicarlo.En el caso de las redes de distribuidores, teniendo en cuenta que las comunicaciones son confidenciales entre el titular y la AEMPS, y que solo los titulares tienen acceso a ellas y a mantenerlas actualizadas, solo es posible cumplir adecuadamente el requisito legal si cada agente realiza sus propias comunicaciones. Solo así podrán cumplir con sus obligaciones sin depender de un tercero.
El nuevo Real Decreto 192/2023 mantiene la obligación de que todos los agentes económicos que comercialicen un Producto Sanitario en España deben comunicar en el Registro de Comercialización a excepción de los puntos de venta exclusiva al público. En este caso la obligación se extiende a todos los productos de cualquier clase, (ver pregunta 98 )
Los productos comercializados a partir de a la entrada en vigor del RD 1591/2009 de productos sanitarios (21 de marzo de 2010) tienen obligación de comunicarse abonando las tasas correspondientes.
Los productos que ya estaban comercializados con anterioridad al 21 de marzo de 2010 deben ser comunicados en un plazo máximo de 6 meses desde la entrada en vigor del nuevo Real Decreto 192/2023 que ha sustituido al RD 1591/2009, sin abono de tasas, según establece la Disposición transitoria primera de dicho real decreto. El plazo de comunicar sin tasa terminó, el 24 de septiembre de 2023, fecha en que todos los productos IIa comercializados en España antes del 21 de marzo 2010 han debido ser comunicados obligatoriamente.
Sí, ya puede solicitarlo porque el justificante de comunicación acredita que ha cumplido con el trámite legal de comunicación. Para obtener el CN debe ponerse en contacto con el Consejo General de Colegios de Farmacéuticos de España. No es competencia de la AEMPS.
Efectivamente, puede comercializarlo. La comunicación de comercialización de un producto con marcado CE es un requisito que se debe cumplir al mismo tiempo en que se comercializa el producto por primera vez en España por esa empresa concreta, y nunca puede ser la comunicación anterior a esta comercialización. La obtención del justificante de comunicación acredita que se ha cumplido el requisito legal de comunicación.
Su comunicación está en estado “Comunicada” y se puede comercializar en España. Los productos sanitarios, en base a su regulación, no se “autorizan” por las autoridades sanitarias.
Las comunicaciones se deben actualizar siempre que haya algún cambio en ellas, (según dispone el RD 1591/2009, artículo 23 punto 2), y se gestionan siempre individualmente, cada una debe tener sus distribuidores actualizados. Si han variado los distribuidores deben modificarlas, por tanto, siguiendo las instrucciones del punto 11.3. Modificaciones múltiples. Seleccione “Distribuidor: Ampliación/Eliminación” si es que desea añadir distribuidores nuevos o eliminar alguno, o bien “Distribuidor: actualización domicilio y/o nombre” si lo que quiere es actualizar sus datos.
El Registro de comercialización del artículo 18 del nuevo RD es obligatorio para todos los agentes económicos de todo tipo que comercialicen en España, solo están excluidas las oficinas de farmacia y los puntos de venta exclusiva al público según se indica en el propio artículo. Entrará en vigor cuando esté operativo, según la Disposición transitoria séptima.
Mientras tanto continúan en vigor los registros nacionales actuales
- Registro CCPS, destinado a productos sanitarios de las clases IIa, IIb y III, (Directiva 93/42/CEE y Reglamento (UE) 2017/745) productos implantables activos (Directiva 90/385/CEE y Reglamento (UE) 2017/745) productos sanitarios para diagnóstico in Vitro de las listas A y B y de autodiagnóstico (Directiva 98/79/EC) y clases B, C y D (Reglamento (UE) 2017/746 consolidado).
- Registro RPS destinado a productos sanitarios de clase I y productos DIV de cualquier clase, comunicados por los fabricantes y representantes autorizados establecidos en España, y a las agrupaciones de productos sanitarios realizadas por sus agrupadores establecidos en España. También para los productos sanitarios a medida por sus fabricantes (establecidos en España y fuera de ella) y por sus representantes autorizados establecidos en España.
Cuando el Registro de comercialización del artículo 18 del nuevo Real Decreto 192/2023. se ponga en marcha (ligado al funcionamiento pleno de Eudamed), todos los productos sanitarios (excepto los productos sanitarios a medida) se comunicarán en la nueva aplicación, y CCPS dejará de estar operativa. Como diferencia reseñable, en el nuevo registro de comercialización, la comunicación debe hacerse antes de comercializar el producto, a diferencia de CCPS en que se comunica en el momento de la comercialización. RPS quedará reservada únicamente a Productos Sanitarios a medida
El Real Decreto 192/2023 no deroga por completo al Real Decreto 1591/2009 ni al Real Decreto 1616/2009, Según la Disposición derogatoria única 1.a) 2º del Real Decreto 192/2023. mantiene en vigor los artículos relativos a las comunicaciones de comercialización en CCPS entre otros. Según la disposición transitoria sexta de este Real Decreto, hasta que Eudamed no sea plenamente operativa, seguirán siendo de aplicación las obligaciones relativas a la realización de las comunicaciones de comercialización y puesta en servicio y el registro de responsables de la puesta en el mercado entre otros. Cuando Eudamed esté plenamente operativa y el nuevo Registro de Comercialización esté disponible se realizarán las comunicaciones previstas en el artículo 18 del nuevo Real Decreto.
El nuevo Registro de comercialización le resulta de aplicación a todos los agentes económicos que comercialicen los productos sanitarios en España, tanto fabricantes que comercialicen directamente en territorio español, como Representantes Autorizados, Importadores y por supuesto Distribuidores que lo hagan. Este registro es distinto del de EUDAMED, aunque toma los datos de éste. Los Fabricantes, igual que los representantes autorizados, importadores y agrupadores, deben comunicar todos sus productos en EUDAMED en base al artículo 31 del Reglamento (UE) 2017/745, independientemente de en qué país concreto se comercialicen. En el Registro de comercialización de España comunican todos los agentes económicos que comercializan los productos en territorio español en base a la facultad que el artículo 30 del Reglamento adjudica a los estados miembros y en España se materializa en el artículo 18 del Real Decreto 192/2023
Reglamentos
La versión 2.01. y superiores de CCPS permite comunicar productos sanitarios y productos sanitarios de diagnóstico in vitro con arreglo a los reglamentos. Para ello, seleccione el botón correspondiente del grupo “Nueva por Reglamento” en el menú de la izquierda.
Para continuar haciendo las comunicaciones con arreglo a directivas, seleccione el botón correspondiente del grupo “Nueva por Directiva” del menú de la izquierda (consulte el manual de usuario CCPS punto 5).
Dependerá de qué regulación cumpla el producto. Si es un producto que tiene un certificado CE válido de directiva y se puede acoger al artículo 120 del reglamento de productos sanitarios, debe comunicarlo como “Nueva por Directiva”. Si, por el contrario, ya está certificado de acuerdo al reglamento de productos sanitarios, debe seleccionar “Nueva por Reglamento”. Lo verá en el tipo de certificado CE que aporte.
Debe recuperar su comunicación seleccionando “Reglamento”, según se explica en el punto 13 del manual CCPS. A partir de ahí aparecerán las pestañas correspondientes al reglamento.
No se pueden mezclar productos de directivas y productos de reglamento en la misma comunicación. Debe desglosar la comunicación en dos, dejando los modelos de directiva en la comunicación actual y haciendo una nueva comunicación acorde a reglamento para los modelos que lo cumplen, abonando la tasa correspondiente a nueva comunicación.
A partir del 26 de mayo de 2021, las agrupaciones deben ajustarse al reglamento, salvo que antes de esa fecha estuvieran legalmente en el mercado UE de acuerdo a la Directiva y cumplan los requisitos del artículo 120 del Reglamento. Si las agrupaciones han sufrido cambios relevantes después de esa fecha, deberán hacer la modificación de “Cambio de legislación a Reglamento”. En cuanto a los componentes, a medida que vayan cambiando de legislación, deben ir haciendo la modificación de cada uno gradualmente. Los cambios quedan recogidos en la comunicación de la agrupación, que puede incluir componentes tanto de directiva y como de reglamento indistintamente.
La casilla “MDR” de la pestaña certificados era una solución provisional mientras CCPS se adecuaba a introducir los productos acogidos a reglamentos. En la versión 2.0.1. de la aplicación ya está disponible la opción de modificación “Cambio de legislación a Reglamento”. Siga las instrucciones del manual de usuario según se explica en la pregunta siguiente. Se recomienda que haga la modificación disponible en estas comunicaciones para normalizarlas con el resto.
Debe realizar la modificación “Cambio de legislación a Reglamento” siguiendo las instrucciones del manual de usuario CCPS, punto 11.2.6. Este cambio implica, aparte de sustituir el certificado por el nuevo correspondiente al reglamento, revisar la clasificación del producto e incluir el número de UDI-DI básico, tanto en el producto como en sus modelos, revisar los desplegables de Categorías, genéricos y subgenéricos y comprobar los etiquetados e instrucciones de uso si han variado. Se recomienda que incluya los códigos NANDO.
Puede consultar el listado de códigos NANDO en la dirección web: https://eur-lex.europa.eu/legal-content/ES/TXT/PDF/?uri=CELEX:32017R2185&from=ES
Si la evaluación de la conformidad de un producto requiere dos documentos emitidos por un ON, si uno se renueva de acuerdo al reglamento, debe renovar también el otro, aunque uno de ellos tuviera una fecha de validez que hiciera pensar que se podría acoger al art. 120 del reglamento de productos sanitarios o al art. 110 del reglamento de productos sanitarios de diagnóstico in vitro. Los procedimientos de evaluación se complementan unos con otros y no es posible mantener procedimientos de dos regulaciones distintas para el mismo producto.
El UDI-DI básico no es obligatorio en las comunicaciones de directiva. Compruebe que está realizando la nueva comunicación con el botón correcto: “Nueva comunicación por Directiva”. En esas pantallas el UDI-DI básico es un dato voluntario. La comunicación se distingue porque aparece (D) a continuación del número. Si ha generado por error la comunicación como de reglamento (botón del menú “Nueva comunicación por Reglamento”) sí que se lo pedirá. Se distinguen por una (R) a continuación del número. Consulte el punto 5 del manual CCPS.
En el apartado “Observaciones” de la modificación puede indicar la fecha inicial de registro de esos modelos acogidos a directiva. En caso necesario, siempre se puede comprobar en el histórico de la comunicación, donde se puede ver la trayectoria de la comunicación a lo largo del tiempo y sus datos en cada momento.
Los productos IVD que eran de autocertificación con la Directiva 98/79/CE y que se reclasifican con la entrada en vigor del nuevo reglamento necesitando la intervención de un ON, si fueron puestos en el mercado antes del 26 de mayo de 2022, pueden acogerse al artículo 110 del Reglamento UE 2017/746, si cumplen los requisitos establecidos en este artículo y tienen unos plazos para obtener el certificado CE, que varían dependiendo de su clasificación. Tendrán que comunicarse en CCPS solo en el momento en que obtengan su certificado CE con arreglo al reglamento, no antes.
Brexit y Suiza
Las comunicaciones afectadas por los cambios de ON, representantes europeos autorizados radicados en el Reino Unido que pasen a ON o representantes ubicados en algún país UE-27 deben ser actualizadas mediante las correspondientes modificaciones múltiples:
- “Actualización certificado CE por cambio de ON (ON Sustitución)”.
- “Representante Europeo Sustitución” Cuando el representante europeo es otro.
- “Representante Europeo: actualización domicilio y/o nombre” cuando el representante europeo es el mismo, pero ha cambiado su domicilio.
En el manual de usuario de la aplicación se describen con detalle estos procedimientos
Por otra parte, las comunicaciones de productos cuyo fabricante está radicado en Reino Unido, a partir del 1 de enero de 2021, deben incluir un representante europeo designado por el fabricante. La AEMPS ha enviado a las comunicaciones afectadas un correo electrónico de aviso para que las actualicen. Encontrará sus comunicaciones en la bandeja “Pendiente Subsanar” con la pestaña “Representante Autorizado” operativa, para que pueda incluir sus datos siguiendo las instrucciones del punto 6.4 del manual de usuario. Esta operación no conlleva pago de tasa. No olvide actualizar la documentación que corresponda y enviar de nuevo la comunicación a la AEMPS una vez introducido el representante para que se conserven los cambios realizados. Hasta que no lo complete no podrá descargar su resumen de comunicación.
El plazo máximo posible para actualizar finalizó el 25 de mayo de 2021, según establece la nota cuyo enlace se indica. Puede consultar el contenido de esta nota en la Nota informativa PS, 20/2020.
Según se indica en la nota informativa publicada en la web de la AEMPS tras la entrada en aplicación del nuevo Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, desde el 26 de mayo de 2021 Suiza no ha renovado el acuerdo de reconocimiento mutuo (MRA, por sus siglas en inglés) UE-Suiza para productos sanitarios.
Como consecuencia de esto, las comunicaciones afectadas deben actualizarse del siguiente modo:
- Productos con fabricante establecido en Suiza: el fabricante debe nombrar un representante autorizado en la UE y las comunicaciones deben actualizarse seleccionando la modificación “Representante Autorizado: Añadir para fabricante de Suiza.”. Es una modificación múltiple que se ejecutará sobre todas las comunicaciones afectadas, independientemente de la actualización de documentación. Esto se explica en el punto 11.3.11. del manual CCPS. Las empresas deben hacer esta modificación que no va a la bandeja “Pendiente de Actualizar Documentación” y deberán aportar las nuevas etiquetas, instrucciones (y certificado CE, si procede) en el momento en que dispongan de esa documentación, a través de la correspondiente modificación individual “Actualización de etiquetas e instrucciones de uso” (esta misma modificación individual permite también actualizar el certificado CE).
- Productos con representante autorizado establecido en Suiza. Deben seleccionar la modificación “Representante Sustitución”. Se explica en el punto 11.3.10. del manual de usuario CCPS.
- Productos certificados por un ON establecido en Suiza. Deben actualizar el Certificado CE. En este caso, el certificado debe estar emitido por un ON establecido en la UE, de acuerdo al Reglamento UE 2017/745 en vigor desde el 26 de mayo de 2021 o al UE/2017/746 desde el 26 de mayo de 2022. Por tanto, la comunicación debe ser transformada de “Comunicación acorde a Directiva“ a “Comunicación acorde a Reglamento“ siguiendo las instrucciones del manual de usuario CCPS. La propia modificación “Cambio de legislación a Reglamento” permite incluir otro ON distinto del anterior.
Todo esto afecta a los productos sanitarios y a los productos sanitarios implantables activos desde el 26 de mayo de 2021, y a los productos sanitarios de diagnóstico in vitro desde el 26 de mayo de 2022.
Información sobre la comunicación de productos sanitarios de diagnóstico in vitro en CCPS
(Nota informativa de la AEMPS PS, 15/2022 de 29 de abril de 2022)
El elemento determinante de la comunicación es el código EMDN hasta un nivel W XX.XX.XX mínimo. Al cumplimentar este campo es posible incluir como modelos distintos productos que tengan en común ese código EMDN, siempre y cuando tengan la misma clasificación, B, C o D. Es necesario que incluyan las etiquetas e instrucciones de uso de todos los productos. Según este sistema, en la pestaña “Producto” son obligatorios solo los campos “Nombre Comercial” (donde deben indicar un nombre genérico que se adapte a los productos incluidos en la comunicación), “Clasificación”, “Nomenclatura EMDN” y “Modelos”. En el apartado “Modelos” es obligatorio el UDI-DI básico y la descripción de cada uno, donde se indicará el nombre comercial real de cada producto. El resto de los campos deja de ser obligatorio, aunque se recomienda su cumplimentación cuando sea posible para permitir búsquedas por el resto de los campos, lo que es útil tanto para la AEMPS como para la propia empresa.
No, el procedimiento mencionado en la nota informativa PS, 15/2022 es solo una opción abreviada de comunicación destinada únicamente a algunos productos DIV de reglamento. Está destinada a facilitar la comunicación a las empresas, teniendo en cuenta el gran volumen de comunicaciones que van a tener que realizar en CCPS para productos que antes eran de autocertificación, y que con el reglamento dejan de serlo. Este procedimiento es opcional y no es el método recomendado por la AEMPS, puesto que disminuye sensiblemente los campos de búsqueda posibles en las consultas. No es aplicable a comunicaciones ya existentes que se recuperan de PMPS, o que ya existen en CCPS y cambian de legislación de directiva a reglamento. Tampoco a DIV que son componentes de agrupación.
El nuevo sistema dificulta las búsquedas de los productos atendiendo a su finalidad de uso y sus características (analito, tipo de muestra, características especiales, etc.) utilizadas por las comunidades autónomas, Cartera de Servicios y otros organismos oficiales para la asignación de concursos y estudios de mercado, y solo se justifica por la especial coyuntura de estos productos, mencionada en la nota informativa PS, 15/2022, hasta la entrada en funcionamiento pleno de EUDAMED.
FAQs de adaptación de CCPS versión 2.0 (Nota informativa de la AEMPS PS, 35/2020 de 16 de diciembre de 2020)
Los nuevos estados equivalentes en CCPS 2.0 son los siguientes:
| Disponibles Nuevas | Comunicadas |
| Disponibles Recuperadas de PMPS | Comunicadas |
| Anotadas | Comunicadas |
| Incidencias pendientes | Comunicadas (pero deben subsanar lo que tengan solicitado por la AEMPS) |
| Incidencias resueltas | Comunicadas |
| Incidencias de Modificación pendientes | Comunicadas pendientes de actualizar documentación |
| Modificaciones pendientes actualizar documentación | Comunicadas pendientes de actualizar documentación |
| Incidencias de modificación resueltas | Comunicadas |
El nuevo documento “Comunicación” es equivalente al antiguo acuse de recibo e indica que el producto ha sido comunicado y, por tanto, es el justificante de que ha cumplido el requisito legal de comunicación, según el RD de PS, de PSIA o de PSDIV, según corresponda.
La Agencia va a realizar la revisión del producto a través de campañas de control de mercado anuales. En dichas campañas se revisará la conformidad del producto con todos los requisitos de la legislación de productos sanitarios, incluida la comprobación de que se ha realizado la comunicación de puesta en el mercado y que dicha comunicación es correcta.
El documento “Comunicación” es el justificante de haber realizado su comunicación y, por tanto, de haber cumplido el requisito legal. Se debe acompañar del resumen de comunicación que puede descargar en cualquier momento de la aplicación y siempre sale actualizado con los últimos datos que haya introducido la empresa.
El acuse de recibo emitido anteriormente por la aplicación es equivalente al actual justificante de comunicación y acredita igualmente el cumplimiento del requisito legal de comunicación. Pero si lo desea, puede descargar en cualquier momento el nuevo modelo de justificante de comunicación, así como el resumen de la misma, que se encuentran en la cabecera de cada una de ellas.
Para adecuarse al nuevo sistema, es necesario que recupere sus comunicaciones desde CCPS y las reenvíe a la AEMPS, actualizando sus datos al mismo tiempo. En ese momento obtendrá su nuevo justificante de “Comunicación” y podrá descargarse su resumen actualizado. PMPS se ha cerrado y no permite ya las consultas desde el 1 de julio de 2020, según se avisó oportunamente en la pantalla de entrada de CCPS.
El procedimiento de recuperación se describe con detalle en el punto 13 del manual de usuario de empresas. Teniendo en cuenta que el 3 de julio de 2018 la aplicación PMPS dejó de estar operativa, la obligación de mantener actualizadas las comunicaciones, y que los certificados CE tienen un periodo máximo de validez de cinco años, el 4 de julio de 2023 no debe haber más comunicaciones en PMPS sin recuperar.
Así se ha informado en la nota PS, 14/2023
Las incidencias pendientes de contestar las verá en el buzón “Pendientes de subsanar” y deberá subsanarlas lo antes posible, igual que en las versiones CCPS 1.5 y anteriores.
Cuando envíe su respuesta a la AEMPS, igual que en la versión 1.5 y anteriores, recibirá un correo informativo de que su respuesta ha sido recibida. Cuando la AEMPS la revise, no recibirá ningún correo si es correcto, puesto que el documento de anotación ya no existe.
Las modificaciones son automáticas e incluyen los nuevos datos sobre la comunicación en el momento de enviarlas a la AEMPS. Las modificaciones que requieren actualización de documentación también son automáticas, pero no vierten los datos hasta completar el proceso. Debe gestionar para ello la bandeja “Pendiente Actualizar Documentación”.
Sí, la aplicación remitirá un nuevo justificante de comunicación con los cambios implementados. En cualquier caso, siempre puede descargar un resumen de comunicación actualizado.
Gestión de las comunicaciones incluidas en las campañas de control del mercado
Es necesario revisar todos los apartados de las comunicaciones incluidas en campaña. Actualice todos los documentos a la última versión de los mismos: certificados, etiquetados e instrucciones de uso. Revise también los modelos incluidos y los desplegables seleccionados en la pestaña “Producto“.
La documentación adicional solicitada en campaña debe adjuntarse en la pestaña “Docs. Asociada“. En la ventana “Observaciones del solicitante“ dispondrá de un cuadro de texto para aclarar cualquier situación que precise. Una vez adjuntada toda la documentación solicitada, es importante pulsar el botón “Enviar doc. a campaña“, ubicado en la esquina inferior izquierda de esa misma pestaña. De esta forma informará a la AEMPS de que ha sido realizada su respuesta.
La aplicación no está diseñada para acusar recibo de una documentación que ha sido requerida en campaña. Como justificante, dispondrá siempre de la visualización de la pestaña “Docs. Asociada“ donde encontrará el listado de la documentación objeto de respuesta y la fecha en la que se realizó dicho envío. Estas comunicaciones permanecerán en la bandeja de “Comunicaciones en campaña“ el tiempo que permanezca abierta dicha campaña.
Los documentos pueden adjuntarse en inglés, pero deben estar correctamente referenciados e identificados en un documento en el que se dé respuesta a la información solicitada de forma concreta.
Si no dispone de esta documentación en el plazo de tiempo otorgado, traslade dicho requerimiento al fabricante o representante autorizado, ya que, tal como viene explicado en el oficio, se dispone de un enlace confidencial para que puedan remitir directamente la documentación a la AEMPS, a través de la opción habilitada en la aplicación CCPS.
No olvide informar al fabricante de la documentación que se le ha solicitado y que debe enviar a través de ese acceso.
Una vez el fabricante o representante autorizado del producto haya sido informado de esta solicitud, usted debe ponerse en contacto con la AEMPS a través del correo electrónico soporteccps@aemps.es solicitando dicha opción. Para ello, deberá indicar en el asunto del correo el siguiente mensaje: ¨Solicitud de token + la identificación de la campaña CAM/XXXX/XXXX¨. En el cuerpo del correo deberá identificar la/s comunicación/es afectadas y confirmar el email al que se enviará dicho enlace. Asimismo, podrá hacer una breve aclaración de esta solicitud a través de la pestaña “Documentación Asociada” en la ventana “Observaciones”.
Si la documentación es enviada por el fabricante o representante autorizado, usted deberá adjuntar la documentación en un plazo de 7 días desde que reciba el enlace de acceso al correo.
El fabricante recibe un correo electrónico con un enlace de acceso a CCPS. Esta página dispone de un desplegable para poder seleccionar el tipo de documento que va a ser adjuntado, como por ejemplo “Análisis de riesgos”, “Datos clínicos”, “Otros” y un botón que indica “Adjuntar”, para poder añadir el documento apropiado. Una vez finalizado dicho paso, debe pulsar en “Enviar”. De esta forma, la AEMPS recibirá de manera confidencial todos los documentos que dan respuesta a la solicitud que le ha sido enviada a usted, como distribuidor del producto en España.
Como ya se informó a través de la nota informativa publicada en diciembre de 2020, se han llevado a cabo los cambios en la gestión de las comunicaciones de comercialización de los productos sanitarios, centralizando su revisión a través de campañas de control de mercado en las que se evaluará en profundidad la documentación técnica del producto y por la cual podrá requerirse toda la información que sea precisa para el desempeño de estas actividades.
Los productos sanitarios de acuerdo con su regulación, no requieren autorización previa de las autoridades sanitarias para su comercialización, si bien tienen que cumplir los requisitos y los procedimientos que figuran en el Real Decreto 1591/2009, de 16 de octubre. Estos productos, dada su clasificación, han sido evaluados por un organismo notificado, quien emitirá dicho certificado y por el cual el producto ostentará Marcado CE.
No obstante, las autoridades competentes efectuarán controles adecuados de las características de conformidad y prestaciones de los productos, pudiendo incluir en su caso, el estudio de la documentación y controles físicos o de laboratorio.
Tal como se indica en el apartado a del punto 3 del artículo 93 del Reglamento 2017/745 del Parlamento Europeo y del Consejo de 5 de abril de 2017 sobre productos sanitarios, las autoridades sanitarias podrán exigir a los agentes económicos, entre otras cosas, que les faciliten la documentación e información que sean precisas para el desempeño de las actividades de las autoridades.
De conformidad con lo establecido en el artículo 120 del Reglamento mencionado anteriormente, los requisitos del Reglamento relativos al seguimiento poscomercialización, control del mercado, vigilancia, registro de agentes económicos y de productos se aplicarán en lugar de los requisitos correspondientes de las citadas directivas.