

La autorización de los medicamentos autorizados por el procedimiento centralizado culmina con la opinión positiva del Comité de Medicamentos de Uso Humano (CHMP, por sus siglas en inglés) de la Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) y la emisión de la decisión de la Comisión Europea. A partir de ese momento, para aquellos medicamentos que se pretendan comercializar en España, se deberá solicitar el Código Nacional (CN).
Autorización inicial
Indicada para solicitar el CN de aquellos nuevos medicamentos centralizados y/o para los cuales nunca se ha solicitado el CN para la misma dosis y forma farmacéutica.
Una vez se haya emitido la opinión positiva del CHMP, y se haya enviado junto a las traducciones de los anexos acordadas con todas las agencias reguladoras la información de producto (ficha técnica, prospecto y etiquetado) al solicitante y a la Comisión Europea (en un procedimiento estándar se refiere a partir del día 237), titular de la autorización de comercialización (TAC) podrá presentar telemáticamente la "Solicitud Cód. Nacional Medicamentos Centralizados" a través de la aplicación RAEFAR II.
Para proceder a dicha presentación, se debe realizar la correspondiente solicitud a través de la pestaña “Comunicación de Autorizaciones de Medicamentos Centralizados (petición CN)” disponible en RAEFAR II, indicando en los distintos campos de la solicitud, tanto el número de procedimiento europeo asignado por la EMA “EMEA/H/ XXXXXX”, como el de registro europeo de medicamento (EU/1/XX/XXXX) y el/los formato(s) a comercializar (EU/1/XX/XXXX/001-005). Es imprescindible rellenar en las pestañas correspondientes y habilitadas a tal efecto en la aplicación cada uno de los formatos que se van a solicitar. Es muy importante que el orden de inclusión de formatos siga el mismo orden de aparición que en el electronic Application Form (eAF).
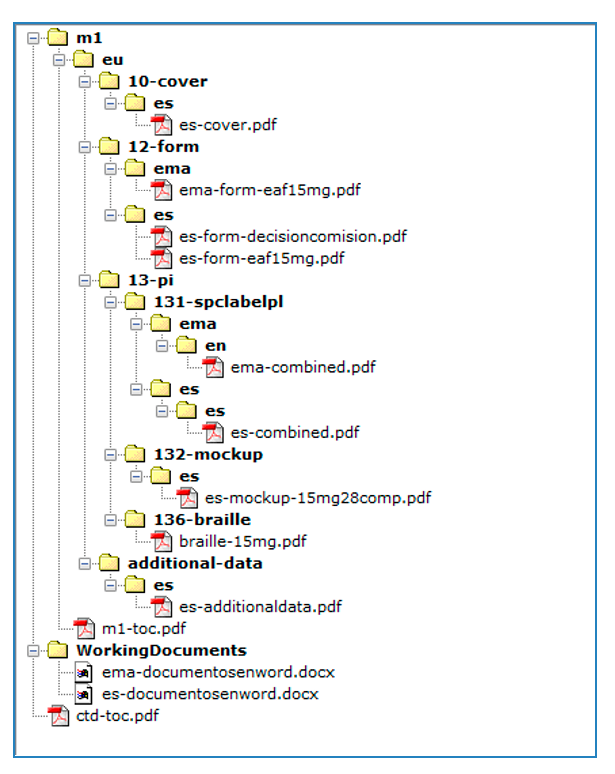
En la secuencia, se incluirá un formulario de solicitud eAF para nuevas autorizaciones de medicamentos que pretenda comercializarse en España, por dosis y/o forma farmacéutica (asegúrese de utilizar la versión en vigor en el momento de la solicitud) disponible para su descarga en la web de Eudralex, o en Esubmission, siendo imprescindible rellenar los apartados marcados como obligatorios para los productos centralizados, (en el caso de, por ejemplo, no estar disponible el código ATC, se indicaría el facilitado provisionalmente). Es de obligado cumplimiento remitir un solo eAF por forma farmacéutica y/o dosis, y poner los formatos asociados a esa forma farmacéutica y dosis, que tengan intención de comercializar en España, en la sección 2.2. Véase esquema en Anexo 1.
En este sentido, el eAF que se ha remitido a la EMA podrá servir para la presentación de la solicitud de CN que se remitirá a la AEMPS, únicamente, en los casos en que el medicamento no haya sufrido modificaciones ni se hayan presentado notificaciones desde su aprobación hasta la presentación de la solicitud del CN, y siempre que los formatos que se incluyan en el eAF cumplan los requisitos mencionados en el párrafo anterior.
En el caso de que durante este intervalo se hayan presentado o aprobado variaciones, o no se cumpla lo anteriormente mencionado en relación a la agrupación por dosis y forma farmacéutica, se deberá cumplimentar un nuevo eAF que se remitirá a la AEMPS para la solicitud del CN.
Los eAFs se incluirán en la carpeta m1/eu/form/es.
- Cover letter, en la cual se indicará el representante local en aquellos casos que proceda.
- Opinión positiva del CHMP y, en caso de disponerse de ella, copia de la decisión de la CE en inglés o en español. En caso de que se solicite el CN antes de la publicación de la decisión, desde la AEMPS se abrirá una solicitud de información adicional en RAEFAR II, para el envío de la misma.
- eAF debidamente cumplimentado, bloqueado y guardado en formato digital.
- Textos en inglés y en español acordados tras la etapa de revisión de comentarios lingüísticos.
- Maquetas aprobadas por la EMA con propuesta de blue box (BB) para su evaluación para aquellas presentaciones que se pretenden comercializar en España. El CN deberá aparecer como XXXXXX.X. En caso de que en el momento de esta presentación el laboratorio no conozca aún el número europeo de registro para cada presentación, este se podrá indicar en la maqueta como EU/1/XX/XXXX/XXX.
- Si procede, el certificado braille correspondiente a dichas presentaciones.
Dicha documentación se presentará adjunta al envío del/de las solicitudes en RAEFAR II, y se estructurará como una Secuencia NEES, incluyendo toda la documentación dentro del Módulo 1. Como anexo 1 a esta instrucción se incluye un modelo de secuencia NEES para realizar dicho envío.
Según el artículo 2.34 del Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente, se define como la persona física o jurídica, normalmente conocida como representante local, designada por el titular de la autorización de comercialización para representarle en España.
Según la legislación comunitaria, se contempla como una figura que es opcional, si bien, para comercializar medicamentos la mayoría de compañías encuentran ventajas en disponer de estructuras locales a través de empresas o entidades que actúan en nombre y representación del TAC.
Al no existir un punto específico en el eAF, en caso de tener designado un RL, deberá indicarse en la cover letter, indicando el nombre completo de la compañía, dirección, teléfono y dirección de correo electrónico de contacto. Con esta información, el RL será incluido dentro de la BBDD de la AEMPS como tal.
En cuestiones relacionadas con la aparición del RL impreso en la información del producto o en la blue box del medicamento, se seguirán las guías y recomendaciones europeas, así como el QRD Template, que se encuentra publicado en la página web de la EMA.
En relación al resto de información necesaria que debe aparecer en la BB, así como su disposición, se encontrarán publicadas las indicaciones necesarias en la página web de la AEMPS.
Si se ha seguido el proceso descrito, la AEMPS enviará en un plazo máximo de 7 días, a contar desde la correcta comprobación de la secuencia con la decisión de la CE en español, y siempre que los materiales sean correctos, la notificación de "Comunicación del Código Nacional para la comercialización en España".
Dicha notificación se recibirá por vía electrónica, mediante un mensaje de aviso a la dirección de correo electrónico que el solicitante indique en los apartados 2.4.1., 2.4.2. y del/de los formulario(s) eAF presentado/s (Titular, Persona designada por él (RL)) como a la Dirección General de Cartera Básica de Servicios del SNS y Farmacia (DGCSF). Dicha comunicación de resolución incluirá el código nacional asignado al formato para el que se solicitó.
De acuerdo con lo establecido en el artículo 28 del RD 1345/2007, el laboratorio titular debe comunicar de como mínimo 15 días antes de efectuarse dicha comercialización. Para señalar la fecha de comercialización efectiva se empleará la aplicación "Notificaciones sobre comercialización de medicamentos".
Asi mismo, se deberá comunicar toda aquella información que sea necesaria previa comercialización del medicamento.
Modificaciones de autorización
Los medicamentos centralizados pueden sufrir modificaciones a lo largo de su ciclo de vida. Si bien estas variaciones son validadas, evaluadas y autorizadas por la EMA, una vez que se autorizan, deben ser comunicadas a nivel nacional para todos aquellos medicamentos que disponen de número de registro y código nacional en España. Las modificaciones que se deben notificar a la AEMPS para su posterior notificación a la DGCSF serán presentadas a través de RAEFAR II.
- Cambio del TAC o la dirección.
- Cambio de representante local o de la dirección.
- Cambio del nombre del medicamento.
- Cambio, supresión o nueva indicación terapéutica.
- Cambio o nueva posología.
- Cambio en las condiciones de prescripción.
- Cambio en el grupo terapéutico (ATC).
- Otros cambios relevantes en la ficha técnica (siempre que afecten a los datos administrativos del medicamento, como es la actualización del liberador de lotes). En el caso de la figura del liberador de lotes, se deberá marcar la opción “liberador de lote” y no será necesaria una secuencia NeeS, si no que únicamente se deberá adjuntar un documento oficial que avale que el liberador de lote es el especificado.
- Cambio de diseño del material de acondicionamiento y/o blue box.
- Comunicación de la anulación de un medicamento/formato (el TAC debe además emplear la aplicación "Notificaciones sobre comercialización de medicamentos").
- Comunicación de la suspensión temporal de comercialización de un medicamento o formato (el TAC debe además emplear la aplicación "Notificaciones sobre comercialización de medicamentos").
- Correcciones de errores.
- Adición de nuevos formatos (nuevos CN): en las tablas del Anexo 1 se muestra un ejemplo de la adición de formatos de medicamentos centralizados.
- Comunicación del levantamiento de la suspensión temporal de comercialización.
La notificación de las modificaciones de los medicamentos centralizados deberá presentarse a la AEMPS para la actualización de los datos del medicamento, una vez que hayan sido autorizadas por la EMA y/o la Comisión Europea.
Serán presentadas a través de RAEFAR II (no siendo necesario incluir un eAF) aportando la documentación necesaria que avale el cambio. Siempre se deberá adjuntar la cover letter, la opinión positiva del CHMP o decisión de la Comisión, y los distintos documentos del medicamento que se vean afectados por el cambio.
Todas aquellas modificaciones que no se encuentren citadas en el listado, serán llevadas a cabo a través de la aplicación de fraccionamiento para la actualización de los textos, siempre que el medicamento se encuentre previamente fraccionado, lo cual es una acción voluntaria en un medicamento aprobado por procedimiento centralizado.
Esta notificación será independiente de la intención del TAC de financiar las modificaciones del medicamento en el territorio nacional.
Si la AEMPS solicita documentación adicional al TAC, este último presentará una secuencia asociada al envío correspondiente.
Habrá 1 envío de Información Adicional por eAF, Dosis y/o FF.
El TAC podrá presentar información adicional o bien a solicitud de la AEMPS o bien antes de que el envío esté “Sin Evaluar”.
Tanto para las Solicitudes de “Autorización Inicial” como para las “Modificaciones de Autorización” encontrará un manual en la sede electrónica de la AEMPS y en la misma solicitud de RAEFAR II.
Una vez recibida la comunicación por la AEMPS, esta procesará la información, actualizará, si procede, la base de datos RAEFAR II, y enviará en su caso, de manera automática, una comunicación a la DGCSF, dando cuenta de este traslado, asimismo, al TAC.
La emisión del oficio de autorización seguirá los criterios internos de la AEMPS, por lo que para algunas modificaciones, puede no ser necesaria la emisión de un oficio si bien RAEFAR II quedará actualizado con la comunicación realizada. Por defecto, se emitirán oficios y se hará traslado a la DGCSF de aquellas modificaciones que se encuentran en negrita en el punto 2.1 de esta guía, aunque por necesidades de la AEMPS o de la compañía, puede encontrarse sujeto a cambios.
Anexos
El TAC solicita el CN para los siguientes formatos (se deberán agrupar para la solicitud si tienen la misma dosis y forma farmacéutica):
| Nº DE REGISTRO EUROPEO DEL MEDICAMENTO | NOMBRE DEL MEDICAMENTO | FORMATOS |
|---|---|---|
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/001 |
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/002 |
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/005 |
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en cartucho | EU/1/XX/XXXX/006 |
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en cartucho | EU/1/XX/XXXX/007 |
| EU/1/XX/XXXX | Denominación 20 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/009 |
El TAC enviará la siguiente documentación a través de 3 envíos (1 por cada eAF):
| Cover | Decisión Comisión | Braille | Nº envíos | Mock-ups | Nº eAFs | Nº Formatos en cada eAF | |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 3 | 1 | 3 | eAF1 | EU/1/XX/XXXX/001 | |
| EU/1/XX/XXXX/002 | |||||||
| EU/1/XX/XXXX/005 | |||||||
| 1 | 1 | 2 | 1 | 2 | eAF2 | EU/1/XX/XXXX/006 | |
| EU/1/XX/XXXX/007 | |||||||
| 1 | 1 | 1 | 1 | 1 | eAF3 | EU/1/XX/XXXX/009 | |
| TOTAL | 3 | 3 | 6 | 3 | 6 | 3 | 6 |
La AEMPS generará en total 3 números de registro y 6 CN ya que los NR AEMPS agrupan los medicamentos en base a un mismo principio activo, dosis y forma farmacéutica, por lo que un NR AEMPS puede agrupar varios NR comunitarios (ver adición de formatos).
| Nº Registro AEMPS | Nº CNs | |
|---|---|---|
| NR AEMPS1 | EU/1/XX/XXXX/001 | |
| EU/1/XX/XXXX/002 | ||
| EU/1/XX/XXXX/005 | ||
| NR AEMPS2 | EU/1/XX/XXXX/006 | |
| EU/1/XX/XXXX/007 | ||
| NR AEMPS3 | EU/1/XX/XXXX/009 | |
| TOTAL | 3 | 6 |
Adicionalmente, una vez autorizados los NR AEMPS arriba descritos, el TAC solicita CN para los siguientes formatos:
| Nº DE REGISTRO EUROPEO DEL MEDICAMENTO | NOMBRE DEL MEDICAMENTO | FORMATOS |
|---|---|---|
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/003 |
| EU/1/XX/XXXX | Denominación 10 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/004 |
| EU/1/XX/XXXX | Denominación 30 unidades/ml solución inyectable en pluma precargada | EU/1/XX/XXXX/010 |
Como ya existe el NR AEMPS1, el formato EU/1/XX/XXXX/003 y EU/1/XX/XXXX/004 se solicitarán como “Modificación de Centralizados”, subtipo “Adición de formatos”, ya que comparten la misma dosis y forma farmacéutica.
Sin embargo, el formato EU/1/XX/XXXX/010 se solicitará como “Comunicación de Autorización de medicamentos centralizados (Petición CN)” puesto que en este caso tendrá una dosis distinta a los formatos que ya tienen CN, con lo que le corresponderá la asignación de un CN y un NR AEMPS distinto.
El TAC enviará la siguiente documentación a través de 2 envíos:
| Cover | Decisión Comisión | Braille | Nº envíos | Mock-ups | Nº eAFs | Nº Formatos en cada eAF | Doc adicional | |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 2 | 1 | 2 | 0 | EU/1/XX/XXXX/003 | 1 | |
| EU/1/XX/XXXX/004 | ||||||||
| 1 | 1 | 1 | 1 | 1 | eAF4 | EU/1/XX/XXXX/010 | 1 | |
| TOTAL | 2 | 2 | 3 | 2 | 3 | 1 | 3 | 2 |
Por lo tanto, visto lo anterior, la AEMPS generará en total 1 número de Registro NR AEMPS4 y 3 CN. Para esos formatos, se emitirán sus correspondientes CN, que quedarán englobados dentro del NR AEMPS1.
| Nº Registro AEMPS | Nº CNs | |
|---|---|---|
| NR AEMPS1 | EU/1/XX/XXXX/003 | |
| EU/1/XX/XXXX/004 | ||
| NR AEMPS4 | EU/1/XX/XXXX/010 | |
| TOTAL | 2 | 3 |
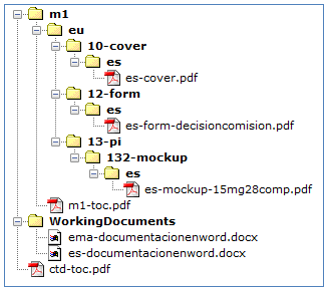
Realizar un envío por número de registro AEMPS a modificar conteniendo todos los formatos (EU/1/XX/XXXX/001-005). Campo de texto libre a completar en RAEFAR:
- Producto y breve descripción de la variación (por ejemplo: nueva indicación, cambio del Código ATC, cambio de TAC, cambio del representante local, etc.).
- Con fecha (día) de (mes) de (año) el laboratorio (titular de la autorización) ha recibido la decisión de la Comisión para la modificación de (motivo de la modificación) para el/los medicamento(s) (nombre(s) del medicamento).
En España dicha modificación afecta a la(s) siguientes presentación(es) de dicho(s) medicamento(s):
- Presentación 1 del Medicamento / Nº de Registro Europeo / C.N.
En apoyo de esta notificación adjuntamos una secuencia NEES incluyendo:
- Copia de la decisión de la CE para dicha variación.
- Copia de la versión vigente de los datos administrativos europeos (obtenida del expediente eCTD del dosier europeo de registros para este medicamento), (si el cambio afecta a los datos administrativos – p. ej.: cambio de código ATC).
- Copia de la información del producto en español emitida con la decisión de la CE (Anexos I-III), (si el cambio afecta a dichos Anexos – p. ej., cambio de indicación).
Igualmente habrá que completar unos campos predeterminados en la citada pestaña de RAEFAR II.