

Última actualización: 15/06/2012
Puede consultar esta nota en formato pdf
*Modificación de 15 de junio de 2012 (ver nota al final)
Fecha de publicación: 24 de febrero de 2012
Categoría: La AEMPS, MEDICAMENTOS VETERINARIOS, INDUSTRIA.
Referencia: MVET, 03/2012
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha adoptado una iniciativa para que, una vez que el Comité de medicamentos Veterinarios (CVMP) de la Agencia Europea de Medicamentos (EMA) haya dictado Opinión Positiva para un medicamento centralizado, el Laboratorio Titular de la Autorización de Comercialización (TAC) pueda con carácter voluntario presentar telemáticamente la «Solicitud de Código Nacional y del Material de Acondicionamiento para la Comercialización en España» a través de RAEVET.
La AEMPS ha venido implantando una serie de mejoras con objeto de agilizar la tramitación de las autorizaciones y modificaciones de comercialización de los medicamentos. Estas mejoras se quieren extender ahora también a los medicamentos que se tramitan por la EMA a través del procedimiento centralizado, cuya autorización culmina con la preceptiva Decisión de la Comisión Europea, y que implica que la AEMPS asigne el código nacional para cada formato, tras la revisión de una parte del material de acondicionamiento, y notifique todo ello al titular de la autorización de comercialización (TAC).
La participación voluntaria del titular en la inclusión de la información de registro en RAEVET, a la cual se adjuntará la solicitud de Código Nacional y del Material de Acondicionamiento, contribuye a disminuir la carga de trabajo administrativo de la AEMPS y permite a su vez integrar la documentación en los nuevos sistemas de notificación automatizada de la Agencia, mejorando así todo el proceso.
El TAC también se podrá acoger a este nuevo proceso de inclusión de información en RAEVET con posterioridad a la Decisión de la Comisión, para cualquier otra presentación ya autorizada por la EMA que pretenda comercializar.
En todo caso, para aquellos medicamentos en los que su titular decida no acogerse a esta iniciativa, la AEMPS seguirá realizando la carga en RAEVET. El titular podrá enviar la «Solicitud de Código Nacional y del Material de Acondicionamiento para la Comercialización en España» después de la Decisión de la Comisión, tal como se viene realizando hasta ahora, es decir, presentando la documentación por Registro General, pero en formato electrónico. A partir de ese momento la AEMPS procederá a revisar el material presentado y notificar la «Comunicación del Código Nacional» para la comercialización en España, y a la carga de las bases de datos.
En consecuencia, los titulares de medicamentos que se tramiten por procedimiento centralizado interesados en seguir la presente instrucción deberán ajustarse a lo siguiente:
(1) AUTORIZACION INICIAL
1.1. Envío de la solicitud
Tras la emisión de la Opinión Positiva del CVMP, el titular podrá presentar telemáticamente la «Solicitud de Código Nacional y del Material de Acondicionamiento para la Comercialización en España» a través de RAEVET. Para proceder a dicha presentación, se debe realizar la carga del formulario de RAEVET para Nuevos Medicamentos, siendo imprescindible rellenar los apartados marcados como obligatorios para los productos centralizados que figuran en negrita y presentar, paralelamente al envío de este formulario, la siguiente documentación:
- Copia de la Opinión del CVMP para dicho medicamento.
- Copia de la versión vigente de los Datos Administrativos Europeos (obtenida del expediente VNeeS del dossier europeo de registros para este medicamento).
- Copia firmada del formulario RAEVET en formato pdf.
- Textos en inglés y en español acordados tras la etapa de revisión de Comentarios Lingüísticos (Día 237 del procedimiento EMA Post-opinión). 1
- Propuesta de las maquetas del embalaje exterior para las presentaciones que se pretenden comercializar en España. El código nacional deberá aparecer como XXXXXX.X. En caso de que en el momento de esta presentación el laboratorio no conozca aún el número europeo de registro para cada presentación, éste se podrá indicar en la maqueta como EU/2/XX/XXX/XXX.
- Si procede, Certificado Braille correspondiente a dichas presentaciones
Dicha documentación se presentará en paralelo al envío del/de los formulario(s) de RAEVET, y se estructurará como una Secuencia VNeeS, incluyendo toda la documentación dentro del Módulo 1. Como anexo 1 a esta Instrucción se incluye un modelo de secuencia VNeeS para realizar dicho envío. El envío de dicha secuencia VNeeS podrá realizarse telemáticamente o en CD.
El formulario de RAEVET se debe completar por cada presentación que se pretenda comercializar en España, es decir por cada número de Registro Europeo (ejemplo: EU/2/11/XXX/001). Una vez completado el formulario para una presentación, se podrá emplear la utilidad de RAEVET «Duplicar solicitud» tantas veces como fuera necesario, modificando en cada formulario únicamente los datos correspondientes a cada presentación.
Para cualquier duda sobre la carga en RAEVET se debe dirigir al correo electrónico “edossierv@aemps.es”.
1.2. Envío de la Decisión de la Comisión
Si la solicitud se hubiera presentado previamente a la Decisión de la Comisión, una vez que el laboratorio obtenga la Decisión de la Comisión para el medicamento, se deberá presentar una nueva secuencia VNeeS con copia de dicha Decisión de la Comisión.
1.3. Notificación de la AEMPS
Si se ha seguido el proceso descrito, la AEMPS enviará en un plazo máximo de 7 días, a contar desde la presentación de la secuencia con la Decisión, y siempre que los materiales sean correctos, la Notificación de «Comunicación del Código Nacional para la comercialización en España».
La AEMPS ha trabajado e implementado el desarrollo informático necesario para que el laboratorio reciba dicha Notificación por vía electrónica, mediante un e-mail de aviso al correo que el Laboratorio indique en los apartados 2.4.1., 2.4.2. y 2.4.6. del/de los formulario(s) RAEVET presentado (Titular, Persona designada por el Titular y Representante local). Dicha Comunicación de Resolución incluirá el código nacional asignado a la presentación para la que se solicitó.
1.4. Comunicación de comercialización efectiva
De acuerdo con lo establecido en el artículo 27 del RD 1246/2008, el Laboratorio Titular debe comunicar de forma expresa a la AEMPS la fecha de comercialización efectiva de cada medicamento (puesta en el mercado del primer lote), como mínimo quince días antes de efectuarse dicha comercialización. Para señalar la fecha de comercialización efectiva se empleará la aplicación «Notificaciones sobre comercialización de medicamentos».2
(2) MODIFICACIONES DE AUTORIZACION
2.1. Notificación de la modificación
Para aquellas modificaciones de los medicamentos centralizados que deban ser notificadas a la AEMPS, tal como se indica en el anexo 2 de la presente instrucción, una vez obtenida la autorización de la autoridad competente, cuando proceda, el laboratorio utilizará la pestaña de RAEVET habilitada para enviar estas notificaciones (SOLICITUD/VARIACIONES/MODIFICACIONES CENTRALIZADOS). En el Anexo 3 se muestra un ejemplo de la información a introducir por cada presentación, a la cual se adjuntará un envío de una Secuencia VNeeS que incluya la documentación pertinente en cada caso (Copia de la Decisión de la Comisión, textos nuevos, si procede, etc).
Para cualquier duda, se deberá dirigir al correo electrónico “edossierv@aemps.es”
ANEXO 1. Modelo de secuencia
VNeeS
para realizar envío del formulario de
RAEVET.
Ejemplo de un envío para 3 presentaciones: 100 comprimidos, 60 comprimidos y 20 comprimidos.
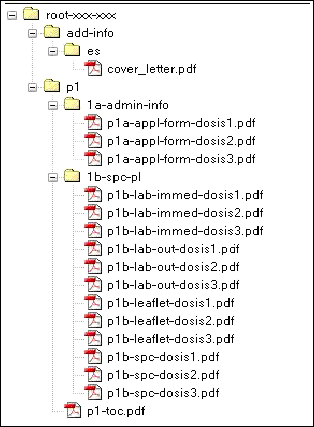
ANEXO 2. Relación de modificaciones que deben ser notificadas a la AEMPS.
- Cambio del Titular o la dirección.
- Cambio de representante local o de la dirección.
- Cambio del nombre del medicamento.
- Cambio, supresión o nueva indicación terapéutica.
- Cambio o nueva posología.
- Cambio en el grupo terapéutico (ATCvet).
- Otros cambios relevantes en la ficha técnica (FT).
- Cambio de diseño del material de acondicionamiento.
- Comunicación de la anulación de un medicamento o Suspensión Temporal de comercialización.
En el caso de la Comunicación de la anulación de un CN o la Comunicación de la Suspensión de comercialización de un CN, además el TAC debe emplear la aplicación «Notificaciones sobre comercialización de medicamentos».3
Para tramitar una nueva presentación de un medicamento ya comercializado en
España se deberá presentar en RAEVET una nueva solicitud de CN con el material
correspondiente y seguir el mismo proceso que se especifica en este
documento.
Asimismo, se deberá comunicará cualquier cambio que lleve implícito la
adjudicación por parte de la AEMPS de un nuevo código nacional.
ANEXO 3. Ejemplo de notificación de modificaciones de la autorización
de comercialización de medicamentos autorizados por procedimiento centralizado
en RAEVET
NOTA: Realizar un envío por presentación (EU/2/11/XXX/001)
CAMPO DE TEXTO LIBRE A COMPLETAR EN RAEVET:
Producto y Breve descripción de la variación (por ejemplo: Nueva indicación,
Cambio del Código ATCvet, Cambio de titular,
Cambio del Representante local, etc).
Con fecha (día) de (mes) de (año) el laboratorio (Titular de la
Autorización) ha recibido la Decisión de la Comisión para la modificación de
(motivo de la modificación) para el/los medicamento(s) (Nombre del
medicamento).
En España dicha modificación afecta a la(s) siguientes presentación(es) de
dicho(s) medicamento(s):
- Presentación 1 del Medicamento / Nº de Registro Europeo / C.N.
En apoyo de esta notificación adjuntamos una Secuencia VNeeS incluyendo:
- Copia de la Decisión de la Comisión para dicha variación.
- Copia de la versión vigente de los Datos Administrativos Europeos (obtenida del expediente VNeeS del dossier europeo de registros para este medicamento) (Si el cambio afecta a los datos administrativos – ej: cambio de código ATCvet).
- Copia de la Información del Producto en español emitida con la Decisión de la Comisión (Anexos I-III) (Si el cambio afecta a dichos Anexo – por ej, cambio de indicación).
Igualmente habrá que completar unos campos predeterminados en la citada pestaña de RAEVET.
ANEXO 4. Instrucciones para los Titulares de la Autorización de
Comercialización y Representantes Locales en lo relativo a los permisos de
inclusión de datos de RAEVET
La inclusión de datos en RAEVET debe ser realizada por el Titular de la Autorización de Comercialización empleando medios propios o autorizando a un tercero que deberá contar con certificado de la Fábrica de Moneda y Timbre para cargar en RAEVET los medicamentos de dicho Titular.
Puede encontrar el formulario de solicitud de acceso y la información
detallada sobre cómo realizar esta solicitud en la dirección de Internet:
https://www.aemps.gob.es/oficinaVirtual/docs/formulario-acceso-Oficina-Virtual.pdf
Instrucciones en relación a los datos de RAEVET que no están dados de alta en las tablas predeterminadas de dicha Base de Datos
Para aquellos datos que no estén dados de alta en las tablas predeterminadas de
RAEVET, tales como principio activo, código ATCvet, Laboratorio Titular, etc;
el Laboratorio deberá enviar un correo electrónico a «edossierv@aemps.es«, adjuntando la
documentación que avale la inclusión de dichos datos en RAEVET (por ejemplo:
datos administrativos europeos y/o copia del establecimiento del titular en la
UE, etc). De esta forma, dichos datos se darán de alta en las correspondientes
tablas predeterminadas, para que el laboratorio pueda seleccionarlos en el
campo correspondiente del formulario de RAEVET a la hora de completar el
mismo.
*Modificación de 15 de junio de 2012: se han corregido los términos
relativos a la nomenclatura de los nombres veterinarios.
- Ver EMA/288844/2009 Rev 4: The linguistic review process of product information in the centralised procedure- veterinary
- https://sinaem.aemps.es/WebComercializacion/login.aspx?opc=veterinaria
- https://sinaem.aemps.es/WebComercializacion/login.aspx?opc=veterinaria