

Preguntas y respuestas frecuentes relativas al Electronic Application Form (e-AF), de aplicación para las siguientes solicitudes:
- Solicitud de Nuevo Registro
- Solicitud de Variaciones
- Solicitud de Renovación Quinquenal
Medicamentos de uso humano y medicamentos veterinarios
- Procedimiento de Reconocimiento Mutuo, Procedimiento Descentralizado y Procedimiento Nacional (PRM / PDC / PN)
Versión 03 (julio 2016)
Índice
- Generalidades sobre el Electronic Application Form
- Cuestiones técnicas
- Envío del expediente electrónico
- Cuestiones comunes a solicitudes de nuevos registros, variaciones y revalidaciones
- Cuestiones específicas relativas a solicitudes de variaciones de la autorización de comercialización
- Varios. Requisitos nacionales
- Glosario de abreviaturas
En caso de tener alguna duda técnica, comentarios o consulta no resuelta en las guías o preguntas frecuentes la EMA dispone de un portal web de soporte donde puede dirigirse.
La URL de acceso es https://servicedesk.ema.europa.eu
Alternativamente para cuestiones puramente nacionales, puede dirigir un correo a soporte_aplicaciones@aemps.es
Nota: Este nuevo portal a través de la web, sustituye a la dirección de correo electrónico eAF@ema.europa.eu utilizada hasta ahora para todas las consultas relacionadas con el e-AF y también reemplaza al buzón de consultas técnicas support@ema.europa.eu
1. Generalidades sobre el Electronic Application Form
El Electronic Application Form es un formulario electrónico único europeo, (que utiliza tecnología PDF para capturar la información y XML para transferirla), preparado para la solicitud de Nuevas Autorizaciones de Comercialización de Medicamentos de Uso Humano y Medicamentos Veterinarios, Variaciones de la Autorización de Comercialización y Revalidaciones.
El Electronic Application Form (e-AF), presenta nuevas posibilidades como la importación/exportación de datos electrónicos, el autocopiado de datos dentro del formulario y la autovalidación del formulario previa a su envío y asimismo proporciona menos campos de texto libre pero si permite el acceso “on line” a catálogos de términos estandarizados (EUTCT).
Dicho formulario será de obligada aplicación a partir del 01 de enero de 2016 para todo tipo de procedimientos de registro, (acorde a la hoja de ruta de la red de Agencias Europeas), aunque habrá un periodo transitorio de dos meses, durante los cuales será necesario rellenar paralelamente el correspondiente RAEFAR/RAEVET.
Nota: El formulario para solicitudes de homeopáticos y otros formularios específicos de la EMA o de las Autoridades Nacionales Competentes (NCAs), por ejemplo el formulario de presubmission o el formulario de notificaciones acordes al artículo 61.3 permanecen con los formatos existentes (se encuentran fuera del alcance del e-AF).
Los diferentes Electronic Application Form están disponibles para su descarga en Esubmission o en la Sede electrónica de la AEMPS
Todas las solicitudes después del 1 de enero de 2016 deberán ser presentadas con el e-AF. Es responsabilidad del solicitante proporcionar la solicitud usando la versión correcta del e-AF.
Desde el 01 de Julio de 2016 la única y última versión del e-AF disponible en la página web es la V 1.20.0.1
(Nota: No será necesario cambiar la versión del e-AF en los procedimientos en curso). Puede obtenerse más información en el siguiente enlace:
http://esubmission.ema.europa.eu/eaf/index.html
Dónde se pueden consultar detalles del plan de fechas para la publicación de las versiones del e-AF:

Fig. 1: eAF versión “Roadmap”
NO. El Electronic Application Form estará disponible únicamente en inglés, acorde al e-Submission Roadmap.
Para procedimientos MRP/DCP, el idioma a cumplimentar en los campos de texto será el inglés.
En el caso de procedimientos nacionales, quedará a elección del solicitante, siendo preferible el español.
SI. ya que no hay traducciones nacionales disponibles. Todos los EEMMs deberán aceptar el formulario de solicitud en inglés como idioma común.
No existen Electronic Application Form disponibles y específicos por país.
No obstante, puede que algunas NCAs requieran un documento adicional y/o traducciones en los documentos de trabajo. Se recomienda contactar con el EMR /NCA correspondiente.
Para las nuevas solicitudes de registro es altamente recomendado el uso de un único e-AF para solicitud de una sola dosis y/o forma farmacéutica.Para los casos de otras dosis (y/o formas farmacéuticas) se presentarán tantos Electronic Application Form completos y separados como dosis / formas farmacéuticas se presenten “For national, mutual recognition and decentralised procedures, a completed separate application form is usually required for each strength and pharmaceutical form”. Ver apartado Purpose and general rules en el documento “USER GUIDE FOR THE ELECTRONIC APPLICATION FORM FOR A MARKETING AUTHORISATION”:
Para las solicitudes de variaciones y/o revalidaciones y para todos los medicamentos afectados por la solicitud, se enviará un único Electronic e-AF.
“In MRP/DCP one common application form is highly recommended, one per pharmaceutical form for all involved products for all member states in case of variations and renewals”.
En el siguiente enlace: http://esubmission.ema.europa.eu/eaf/index.html se encuentran documentos tales como:
Guidance Documents.-Regulatory:
User Guide for the Electronic Application Form for a marketing authorisation (Human)
User Guide for the Electronic Application Form for a marketing authorisation (Veterinary)
(Guías que facilitan el trabajo de los solicitantes a la hora de cumplimentar los e-AF, como parte de una solicitud de autorización de comercialización de un medicamento de uso humano / medicamento veterinarios)
Guidance Documents.-Technical:
(Proporciona un apoyo práctico y técnico sobre el uso de los e-AF para medicamentos de uso humano y veterinario)
Nota: Se recomienda consultar las Guías regulatorias y técnicas en paralelo:
Questions and Answers (FAQ´s): User Guidance Human & Vet http://esubmission.ema.europa.eu/eaf/docs/eAF%20Question%20and%20Answers%20-%20update.pdf
También se recomienda consultar el siguiente documento de utilidad:
Electronic Application Form training for industry: Webinar for industry – Use of Human and Veterinary e-AF http://esubmission.ema.europa.eu/eaf/docs/eAF%20training%20Jan%202016%20industry(2).pdf
2. Cuestiones técnicas
Se deberán revisar las infraestructuras tecnológicas con el departamento de Informática correspondiente, (IT), ya que las especificaciones mínimas para trabajar correctamente con el e-AF son Adobe Reader/Acrobat 10 o Adobe Reader/Acrobat 11 (siendo recomendable siempre utilizar la última versión).
Para más información consultar la sección “Requirements on Adobe Reader and IT security settings” en el document “Harmonised Technical Guidance for Using of Electronic Application Form for human and veterinary medicinal products in the EU” Se deberá comprobar asimismo la conectividad de internet a EUTCT: The European Union Telematics Controlled Terms System (ej. para búsqueda de sustancias activas, formas farmacéuticas, especies de destino, …)
IMPORTANTE: Esta ubicación de servicios web, gestionada por la EMA, permite búsquedas y listas desplegables que se rellenan de forma dinámica en muchos de los campos de los formularios.
Sin acceso a EUTCT el formulario NO se puede completar
El visor de archivos PDF que utilizan Mozilla Firefox y Google Chrome no soporta XFA basado en PDF.
Para solucionarlo pueden consultar el siguiente documento de ayuda de Adobe
http://helpx.adobe.com/livecycle/kb/xfa-forms-firefox-chrome.html
Al guardar el e-AF con Adobe Acrobat deberá hacerlo siempre en formato PDF 1.5 o superior, ya que las anteriores versiones 1.3 y 1.4 no soportan XFA y hacen que el formulario no pueda mostrarse correctamente con el visor.
Cuando se trate de e-CTD, deberán asegurarse que la línea «Document processing” está marcada (ver campo “subrayado” en siguiente imagen) para permitir el manejo del índice de texto completo, incluyendo los catálogos, acorde a los criterios de validación.
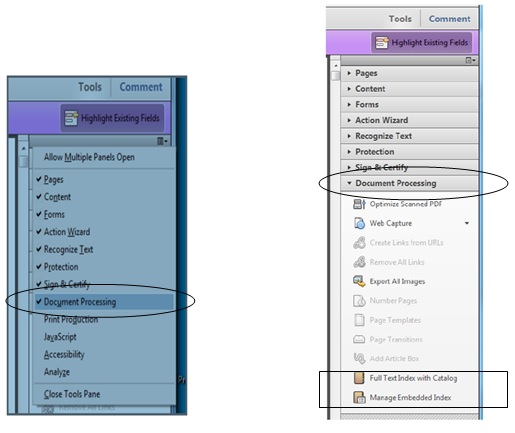
Fig. 2: Configuración Adobe
El e-AF tarda más en abrirse que otro documento de PDF o Word debido a que necesita estar permanentemente conectado a un servicio web, de manera que al abrirse, se cargan las listas desplegables desde EUCT y las correspondientes políticas de cumplimentación que hacen que el formulario sea pesado.
Los campos de búsqueda no funcionan si no está conectado a Internet (sustancias activas, excipientes, códigos ATC…), pero hay algunas listas desplegables que se cargan al abrir por primera vez el formulario y que hacen que tarde más en abrirse.
El tiempo medio que tarde en abrirse dependerá del formulario: no tardará igual cuando se abra por primera vez desde la web, que al abrir uno ya relleno, bloqueado y enviado por el solicitante.
Fig. 3: Mensaje de advertencia
El e-AF necesita estar en comunicación con el servidor (host:“esubmission.ema.europa.eu”). Pinche sobre Opciones/Agregar “host” a Ubicaciones Privilegiadas o vaya a Seguridad Mejorada/Preferencias.
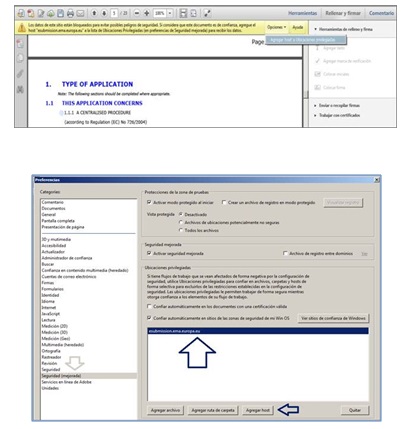
Fig. 4: Configuración seguridad Adobe
El uso del e-AF no modifica los requisitos nacionales respecto a la firma, y seguirá siendo la misma persona la que firme como lo hacía anteriormente (AF Word). Para bloquear el formulario se introducirá la firma escaneada a modo de imagen.
Esta firma es únicamente informativa, sin validez legal, pero necesaria para bloquear el formulario y hacerlo consistente y seguro, permitiendo la posterior importación/exportación XML y asegurando que no se modificará la información introducida por el solicitante.
El formulario debe guardarse con formato electrónico, evitando imprimir y escanear el PDF porque pierde su funcionalidad, y además sería motivo de rechazo por las Autoridades Nacionales Competentes (NCAs).
3. Envío del expediente electrónico
3.1. Cuestiones comunes a solicitudes de nuevos registros, variaciones y revalidaciones
El envío de dichos formularios se realizará junto a la secuencia correspondiente a través de la plataforma europea CESP.
El envío será único y lo realizará el solicitante para el EMR y todos los Estados Miembros Concernidos (EMCs), y por medio de la plataforma CESP se lanzará el envío simultáneamente a todos los países.
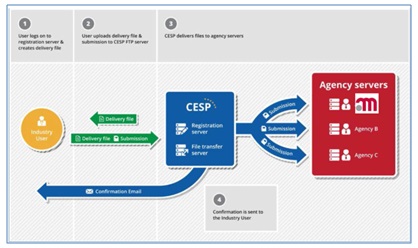
Fig. 5: CESP
Es altamente recomendable enviar un formulario común y único en inglés como lengua común y no habrá que remitir traducción del e-AF en español con la secuencia.
En el caso de medicamentos de uso humano:
El archivo e-AF debe ser incluido en la secuencia e-CTD/NEES de la solicitud en la carpeta.
“m1eu12-formcommon” nombrado como “common-form-partevariable.pdf”
En el caso de medicamentos veterinarios el archivo e-AF deberá ser incluido en la secuencia VNeeS de la solicitud en la carpeta:
“p11a-admin-info” nombrase por ejemplo como “p1a-application-form.pdf”
NOTA: Se recuerda que siempre se debe enviar el e-AF dentro de la solicitud (eCTD / NeeS / VNeesS). No se deberá presentar el e-AF fuera del dossier.
La firma puede incluirse por el solicitante o por un representante autorizado, ya que la política actual no ha cambiado en el e-AF (se recomienda añadir un poder notarial o legal como se requiere por los EEMM).
Lo ideal sería proporcionar un único punto de contacto. Para aquellas NCAs que requieren múltiples personas de contacto se incluirá un anexo separado con los datos de contacto.
El uso del e-AF será obligatorio para todas las solicitudes, independientemente del formato de solicitud.
Dado que la información en las secciones 2.1, 2.2, 2.4 y 2.6, así como en los documentos de apoyo anexados y en el apartado “Declaración y Firma” debe ser idéntica, cuando se completa este apartado, los datos se “autocopian” a los campos correspondientes de otras secciones del e-AF, marcando la opción “Populate data” (push-button) Así, por ejemplo, valores como los campos “Pharmaceutical form”, “Strength” and “Active Substance” son autocopiados de la sección “Administrative data”.
El e-AF proporciona menos campos de texto libre, pero sí permite el acceso “on line” a catálogos de términos estandarizados (EUTCT). Así, mediante la opción “seleccionar listas desplegables (“drop-downlist”) aparecen listas de términos / vocabularios controlados, seleccionados de catálogos EUTCT.
Las listas desplegables incluyen términos acordes a nomenclaturas estandarizadas y a estándares de calidad para sustancias y para medicamentos del EDQM o del sistema EUTCT.
También se describen funciones de las entidades en los desplegables, como se puede ver a continuación en los ejemplos:
A brief description of functions performed for each manufacturer(s) concerned is available in the dropdown field based on a controlled dictionary (e.g., Primary packaging, quality control testing, Processing of medicinal product, …)
A brief description of manufacturing steps performed by manufacturing site for each manufacturer(s) concerned is available in the dropdown field based on a controlled dictionary (e.g., Manufacture of active substance, Manufacture of active substance intermediate, Packaging of active substance, …)
Nota: Si los términos no están disponibles en los campos desplegables, se deberán solicitar a través del formulario “New term request”, por correo electrónico dirigido al Master Data Management Service (MDMS).
- New term requests: mdms@ema.europa.eu
(Importante: Utilizar este correo siempre para solicitar nuevos términos. NO utilizar el correo eAF@ema.europa.eu, porque se puede retrasar la gestión de la solicitud).
Debido a la posibilidad de tener diferentes anexos adjuntos, es altamente recomendable seguir la convención de nombres (Extendiendo el uso de la parte variable): eCTD y NeeS/VNeeS.
Todos los archivos anexados deberán hacer uso de la parte variable para identificar diferentes contenidos (por ej. Anexo de la sección 2.6.2., comprobante de pago de tasas o anexo 5.19):
- cc-form-annex2-6-2.pdf
- cc-form-proofpayment.pdf
- cc-form-annex-5-19.pdf
Para VNeeS, los anexos se incluirán en la carpeta XXXXp11a-admin-info; al no existir una convención detallada de nombres se recomienda nombrar los archivos para que semánticamente se puedan asociar al anexo que corresponden, por ejemplo:
- p1a-annex-5-1-proof-of-payment.pdf
- p1a- form-annex-5-19.pdf
- p1a- form-annex-2-1-2.pdf
3.2. Cuestiones específicas relativas a solicitudes de variaciones de la autorización de comercialización
Se presentara como hasta ahora, si era NeeS en NeeS si era en e-CTD en e-CTD. Con la salvedad que ya conocéis: “e-CTD: Único formato aceptado en procedimientos centralizados, así como en las nuevas solicitudes para autorización de comercialización de medicamentos de Uso Humano por procedimiento descentralizado (desde junio 2015).
Nota: para medicamentos veterinarios se seguirá presentando por VNeeS.
Ya existe este cambio pendiente para mejorar la solicitud en próximas versiones.
Dado que es un cambio importante en el formulario y debido a la complejidad del cambio propuesto, durante este tiempo se recomienda lo siguiente:
- ¿Cómo añadir la pagina o páginas de la Guideline, con la variación marcada (
 )?
)?
Se añadirá como documento anexado al Electronic Application Form. Deberá incluirse en la secuencia, en la misma ubicación del e-AF y claramente nombrado como “Classification Guideline”.
- ¿Cómo marcar las condiciones a cumplir y la documentación necesaria para cada variación especifica?
Se añadirá como documento anexado al Electronic Application Form. Deberá ser incluido en la secuencia, en la misma ubicación del e-AF, claramente nombrado como “Classification Guideline”.
Es importante no adjuntar “como archivo” los anexos al e-AF, para prevenir el bloqueo automático del mismo.
En la versión vigente del e-AF de variaciones se incorporaron todas las variaciones descritas en la Guideline de variaciones y además una serie de variaciones «z» (muchas procedentes de las recomendaciones del Artículo 5).
Ver e-AF Release Notes: In Section 3 – Scope list has been revised and z-scope has been added in relevant sections
Las descripciones más complejas deberán ser incluidas también en un anexo, la misma ubicación del e-AF claramente identificado.
En principio sería aceptable un solo e-AF en inglés y firmado únicamente por el responsable regulatorio global.
La firma principal confirmará estar autorizada para firmar en nombre de los contactos designados, como se especifican en la sección 2.4.3 de la Parte IA, Modulo 1 del e-AF para cada una de las AC concernidas.
Para solicitudes de Worksharing (y Supergrupos) con más de una AC, se indicará el titular de la autorización de comercialización (TAC) que va a actuar como titular de referencia para el manejo del procedimiento.
En cuanto al apartado Signature y el formato técnico que debe llevar (“los requerimientos se referirán a una «high quality scanned signature image file») se recomienda consultar los documentos: “USER GUIDE FOR THE ELECTRONIC APPLICATION FORM FOR A MARKETING AUTHORISATION” y el Electronic Application Form– Questions & Answers”
El formulario existente en papel “CMDh STANDARD OPERATING PROCEDURE PROCEDURE FOR ARTICLE 61(3) CHANGES TO PATIENT INFORMATION” y disponible en la página web del CMDh.
NO ha sido sustituido, y no está previsto reemplazar esta plantilla en un futuro próximo. Se deberá adjuntar como anexo al formulario de solicitud.
4. Varios. Requisitos nacionales
Ver nota informativa SG, 3/2015: https://www.aemps.gob.esinforma/notasinformativas/industria-2/2015/ni-sg_03-2015-ampliacion-plazo-tasas
Donde se incluyen aclaraciones al plazo de 10 días entre la fecha del pago de la tasa y la presentación del servicio para los procedimientos nacionales.
-
NUMERO DE JUSTIFICANTE:
- Para solicitudes de nuevos registros NO se indicará el número de justificante en el e-AF.
-
En el caso de las solicitudes de variaciones/revalidaciones, será necesario incluir el número de justificante de pago de tasas en el e-AF:
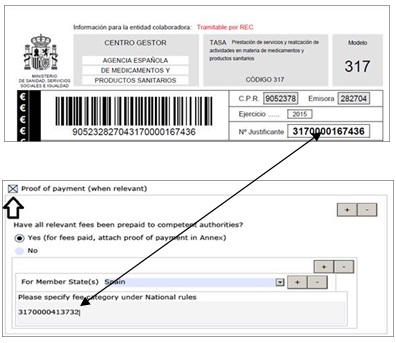
Fig. 6: TasasEn el caso de tener que añadir tasas complementarias, se introducirán separados por punto y coma en el mismo recuadro.
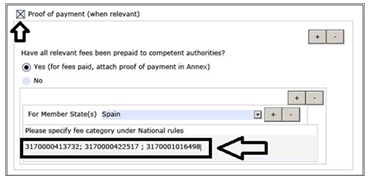
Fig. 7: Tasas complementarias
-
INCLUSIÓN DEL JUSTIFICANTE EN LA SECUENCIA:
Para solicitudes de nuevos registros se incluirá:
-
Para medicamentos de uso humano
En el caso de MRP y DC, se incluirá en la carpeta “common”, si se trata de un justificante común o documento que recoja todas las tasas pagadas:
- \m1\eu\12form\common\common-form-proofofpayment.pdf
- Si los justificantes son por país habrá que incluir cada uno en su carpeta nacional
En el caso de Nacionales se incluirá en la carpeta “form-es”
- \m1\eu\12form\es\es-form-proofofpayment.pdf
- Para medicamentos veterinarios en «add-info» de la carpeta «root» o directamente como anexo en 1a-admin-info, de la carpeta p1.
Para solicitudes de variaciones/revalidaciones se incluirá:
-
Para medicamentos de uso humano
En el caso de MRP y DC, se incluirá en la carpeta “common”, si se trata de un justificante común o documento que recoja todas las tasas pagadas:
- \m1\eu\12form\common\common-form-proofofpayment.pdf
- Si los justificantes son por país habrá que incluir cada uno en su carpeta nacional
En el caso de Nacionales se incluirá en la carpeta “form-es”
- \m1\eu\12form\es\es-form-proofofpayment.pdf
- Para medicamentos veterinarios en «add-info» de la carpeta «root»
-
Las transferencias no entran dentro de la categorización de la Directriz de Variaciones, aunque hasta ahora se trataban como Variaciones tipo II, calendario de 60 días (para los procedimientos europeos era el mismo sistema pero cargadas como ZZ).
A partir de la publicación del e-AF, SOLAMENTE se pueden volcar en el nuevo RAEFAR/RAEVET exclusivamente las variaciones que están contempladas en dicho formulario.
Por este motivo todo lo que no se pueda cargar con el nuevo sistema, se hará de forma independiente en una nueva «pestaña de solicitudes de transferencias” (de titularidad y/o de representante local).
Para los casos en los que la transferencia vaya unida a una solicitud de cambio de denominación del medicamento, este último cambio se solicitará por la vía e-AF, sea el procedimiento nacional o europeo (RM/DC).
En un breve plazo de tiempo la AEMPS publicará instrucciones relacionadas con este tipo de solicitudes de transferencias de titularidad.
SI, esta aplicación seguirá en paralelo como actualmente, realizándose por la aplicación Gestión telemática de Ficha Técnica y Prospectos disponible para su descarga en la sede electrónica de la AEMPS.
Ver enlace:
https://sede.aemps.gob.es/usoHum/regMed/gestion-telematica-ficha-tecnica-prospecto.htm
Para los PRM/PDC, al tratarse de la fase nacional de traducciones, el fraccionamiento en la fase nacional de traducciones también seguirá igual que en la actualidad.
La información adicional para todo lo que está en marcha, en validación y/o evaluación se podrá presentar vía CESP o vía Pestaña “Histórico- Información adicional”.
En estos casos, se marcará la casilla“National authorization in MRP/DCP”
Y en el campo “Variation Procedure Number” indiquen ZZ/H/NNNN/IA, IB o II (según proceda)/nnnn
- Siendo: NNNN el núm. de procedimiento europeo)
- Siendo nnnn el núm. secuencial que le corresponda a la solicitud (0000 = nuevo y si no el correlativo al que se haya asignado previamente a otro envió ZZ en el e-CTD/NeeS)
No existe punto especifico, como en el anterior AF, para indicar el representante local, por lo que se indicará en la “cover letter”, y desde Validación, se procederá a grabar en Raefar dicho punto, con los datos facilitados por el solicitante.
Tendrá que remitirnos nueva secuencia con eAF corregido, siendo importante, que indique en el formulario “Procedure Number”, el número de procedimiento ya asignado por la AEMPS, bien accediendo a Reafar II o en el asunto del correo recibido, una vez cargada en la Base de Datos.
En el caso de nuevos medicamentos y variaciones nacionales (también revalidaciones) al no estar disponible en número de procedimiento se indicará N/A.
En el caso de procedimientos MRD/DCP es importante cumplimentar correctamente dicho número según el patrón común establecido, tanto para nuevos medicamentos, como para variaciones.
Solo en el caso, que hubiera que remitir nuevo eAF, por petición expresa de la AEMPS, indicaríamos ya el número asignado por Raefar II.
En ese caso, habría que incluir tantas cajas de situación actual y propuesta como variaciones vaya a solicitar, indicando la tipificación correcta en cada caso.

Fig. 8: Variaciones eAF 1, Fig. 9: Situación Actual/Propuesta eAF
Glosario
- AASS: Autoridades Sanitarias
- AC: Autorización de Comercialización
- AEMPS: Agencia Española de Medicamentos y Productos Sanitarios
- CE: Comisión Europea
- CMDh: Grupo de Coordinación para Proc. de Reconocimiento Mutuo y Descentralizado, (Medicamentos de uso humano)
- CMDv: Grupo de Coordinación para Proc. de Reconocimiento Mutuo y Descentralizado, (Medicamentos Veterinarios)
- CTD: Common Technichal Document
- e-CTD: Versión electrónica del CTD
- EDQM: European Directorate for the Quality of Medicines & HealthCare
- EEMMs: Estados Miembros (de la Unión Europea)
- EMA: Agencia Europea de Medicamentos
- EMC: Estado Miembro Concernido
- EMR: Estado Miembro de Referencia
- EUTCT: The European Union Telematics Controlled Terms System
- HMA: Heads of Medicines Agencies (Jefes de Agencias de Medicamentos)
- MAA: Marketing Authorisation Application (solicitud de autorización de comercialización)
- NeeS: Non-eCTD Electronic Submissions (Medicamentos de uso humano)
- NTA: Notice to applicants
- PC: Procedimiento Centralizado
- PDC: Procedimiento Descentralizado
- PN: Procedimiento Nacional
- PRM: Procedimiento de Reconocimiento Mutuo
- TAC: Titular de la Autorización de Comercialización
- VNeeS: Non-eCTD Electronic Submissions (Medicamentos veterinarios)