

Versión 7
En estos momentos hay una limitada disponibilidad de las nuevas alternativas terapéuticas antivirales frente a la infección por SARS-CoV-2. Esto hace necesario establecer unos criterios de priorización en el acceso precoz a los mismos.
Las alternativas disponibles que se consideran en este documento son (Anexo I).
- Antivirales
- Veklury (remdesivir)
- Paxlovid (nirmatrelvir/ritonavir)
- Lagevrio (molnupiravir)
- Anticuerpos monoclonales
- Ronapreve (casirivimab / imdevimab)
- Xevudy (sotrovimab)
- Evusheld (cilgavimab /tixagevimab)
La accesibilidad a los tratamientos disponibles se ha priorizado considerando aspectos como la gravedad y/o el riesgo de complicaciones, la respuesta a los tratamientos disponibles y el beneficio esperable.
Algunos de los tratamientos indicados en este documento son moléculas de nuevo desarrollo y otros son medicamentos ya autorizados. De todos ellos se genera constantemente nueva información, que debe ser analizada conforme está disponible para la toma de decisiones por las agencias reguladoras. Además, es necesario actualizar e informar del procedimiento establecido para que se pueda acceder a los medicamentos disponibles. Es por ello que este documento está sometido a actualizaciones continúas conforme se disponga de nuevas evidencias científicas, según la evolución de la epidemia y según la evolución de los stocks de medicamentos disponibles. La AEMPS cuenta para ello, con un grupo de expertos formado por representantes de sociedades científicas y de comunidades autónomas (Anexo II).
La Agencia recomienda a los profesionales sanitarios que consulten las fichas técnicas de los medicamentos que estén autorizados en lo relativo a las recomendaciones de dosis, advertencias y precauciones, posibles reacciones adversas e interacciones.
El acceso a estos medicamentos se realizará por los cauces habituales, en el caso de Evusheld, la distribución se realizará por los cauces ya establecidos. En aquellas CC. AA. que no dispongan de un depósito estratégico de anticuerpos monoclonales (Xevudy, Ronapreve), así como aquellas que lo indiquen expresamente, la solicitud de acceso individual para cada paciente se podrá hacer a través del portal de medicamentos en situaciones especiales (MSE) de la AEMPS. En la mayoría de los hospitales, esa solicitud se gestiona telemáticamente desde el Servicio de Farmacia a partir de la indicación del médico y el visto bueno de la dirección médica (que en algunos hospitales puede no ser requerido de forma individual en las condiciones de priorización descritas a continuación). La gestión debe realizarse de forma inmediata tras la indicación médica y la respuesta de la AEMPS se debe recibir en 24 horas.
En el caso de Paxlovid, debido al perfil de interacciones y advertencias especiales de uso, se requiere una validación farmacéutica previa a su dispensación. Este circuito de validación lo establecerá cada CC.AA. y debe ser inferior a 24h. En el caso de pacientes privados y pertenecientes a MUFACE, MUGEJU e ISFAS con asistencia sanitaria privada, el acceso a Paxlovid se tramitará a través de MSE.
En el caso de los anticuerpos monoclonales, siempre que sea posible, se debe discriminar previamente cual es la variante que está produciendo la infección. La decisión sobre el uso de anticuerpos monoclonales debe tener en cuenta lo que se conoce sobre las características de los virus del SARS-CoV-2 circulantes y la información disponible sobre los patrones de sensibilidad a los mismos.
Se recomienda consultar la última información disponible al respecto:
- https://covdb.stanford.edu/page/susceptibility-data/
- https://opendata.ncats.nih.gov/variant/activity
En el caso de los anticuerpos monoclonales, estos han mostrado una mayor eficacia en pacientes con serología negativa. Por tanto, es necesario determinar el estado serológico antes de su prescripción. Con el objetivo de incorporar más pacientes que se puedan beneficiar del tratamiento con anticuerpos monoclonales se ha considerado que los pacientes con un bajo nivel de protección también pueden beneficiarse de este tratamiento.
Hasta ahora se ha establecido que si el título de anticuerpos frente a la proteína S es menor de 260 BAU/ml se considera una respuesta inadecuada a la vacunación. Este umbral se estableció a partir de un estudio realizado en el contexto de la cepa original de Wuhan, al igual que las instrucciones de uso del Panel Internacional de Normas de Referencias de la OMS para anticuerpos anti-SARS-CoV que se establecieron en diciembre 2020. Hasta la fecha no se ha validado el nuevo umbral frente a las diferentes variantes de interés, por lo que actualmente este umbral se puede considerar orientativo e indicativo de una pobre respuesta a la vacunación. Por otro lado, para demostrar una respuesta adecuada sólida a la vacunación sería necesario disponer de serologías seriadas que aseguraran el mantenimiento en el tiempo de la respuesta inmune lo cual no es posible desde el punto de vista práctico.
Por todo ello, además de valorar la cuantificación de los títulos de anticuerpos frente a la proteína S, se propone tener en cuenta el grado de inmunosupresión del paciente. Es decir, la interpretación del resultado de la serología tendrá que llevarse a cabo junto con las características del paciente en cuanto a su grado de inmunosupresión y el riesgo individual de infección.
La decisión de considerar a la población elegible para este tratamiento tiene que basarse prioritariamente en los resultados de serología (anticuerpos frente a proteína S).
Se recuerda la importancia de notificar todas las sospechas de reacciones adversas al Centro Autonómico de Farmacovigilancia correspondiente o a través del formulario electrónico disponible en www.notificaram.es
Criterios
La intención del documento es identificar criterios de priorización para el acceso precoz a las nuevas alternativas terapéuticas antivirales considerando su disponibilidad limitada. No debe ser considerada en ningún caso como una recomendación de uso por parte de la Agencia.
El acceso y utilización de los medicamentos ya disponibles en sus indicaciones autorizadas debe realizarse siguiendo los protocolos de manejo clínico de cada hospital.
Paciente grave o crítico
Adultos o adolescentes mayores de 12 años con >40kg de peso hospitalizados por COVID-19 grave o crítico que presente serología negativa para SARS-CoV-2, o con bajo nivel de protección para SARS-CoV-2.
En este contexto, el tratamiento con anticuerpos monoclonales se restringe a los pacientes con las condiciones de alto riesgo descritas más adelante (Condiciones de alto riesgo priorizadas).
Debido a la gravedad de los pacientes, no es necesario considerar una ventana de inicio del tratamiento. Se seleccionará el anticuerpo monoclonal (Ronapreve, Xevudy o Evusheld) en función de la disponibilidad, situación epidemiológica local y actividad in vitro. Siempre que sea posible, se debe identificar la variante y tener en cuenta los datos más recientes disponibles.
En ambos casos, se trata de una indicación del medicamento no autorizada.
Paciente de alto riesgo y enfermedad leve-moderada
Estos pacientes, siempre y cuando presenten las condiciones de alto riesgo descritas en el presente documento, pueden ser candidatos tanto a anticuerpos monoclonales (Ronapreve, Xevudy o Evusheld) como a antivirales (Remdesivir, Paxlovid, Lagevrio).
Sin embargo, por su eficacia, facilidad de acceso y uso, se considera que la primera opción terapéutica recomendada es Paxlovid (nirmatrelvir/ritonavir) en pauta de 5 días. Se debe iniciar tratamiento dentro de los 5 días de evolución. Se resalta la importancia de valorar previamente interacciones farmacológicas.
En el caso de los pacientes incluidos en el apartado 1 que no sean candidatos a Paxlovid, si se dispone de resultado serológico que muestre que no hay respuesta a la vacunación (serología negativa o nivel de protección bajo) puede considerarse la administración de anticuerpos monoclonales.
En el resto de los casos (apartado 1 si no se dispone de serología o se ha confirmado una respuesta positiva a la vacuna, así como los apartados 2, 3 y 4), se puede considerar como alternativa el uso de:
- Veklury (remdesivir) en pauta de 3 días. Se debe iniciar tratamiento dentro de los 7 días de evolución.
- Lagevrio (molnupiravir), en pauta de 5 días. Iniciar tratamiento dentro de los 5 días de evolución.
En el paciente de alto riesgo con enfermedad leve-moderada, tanto los anticuerpos monoclonales como los antivirales se utilizarán de acuerdo a las condiciones de la ficha técnica autorizada.
El uso de Paxlovid en los pacientes con replicación viral persistente sintomáticos debe valorarse de manera individualizada por el centro hospitalario y en consenso con la Comunidad Autónoma.
Consideraciones en el paciente pediátrico
De entre las nuevas alternativas para el tratamiento de la infección por SARS-CoV-2 contenidas en el este documento, la opción terapéutica elegible para pacientes menores de 12 años, basado en la evidencia disponible, debería ser remdesivir. Debería considerarse su uso en pacientes menores de 12 años con los criterios de riesgo descritos en este documento, que presenten sintomatología respiratoria leve/moderada motivada por el COVID-19, con alto riesgo de empeoramiento de su infección viral, en el contexto de su vulnerabilidad de base. En los casos de hallazgo de COVID-19 en un contexto clínico sin los criterios previos, de aparición nosocomial sin sintomatología respiratoria, se deberá monitorizar la posible evolución de la infección viral con una determinación seriada de PCR y evolución del número de ciclos.
En caso de agravamiento posterior de un caso, tras remdesivir debería valorarse la utilización de líneas terapéuticas diferentes de los anticuerpos monoclonales, con mayor evidencia que estos en el momento actual.

Condiciones de alto riesgo priorizadas en adultos
1. Personas inmunocomprometidas y con otras condiciones de alto riesgo, independientemente del estado de vacunación:
- Receptores de trasplante de progenitores hematopoyéticos o CAR-T, en los dos años tras el trasplante/tratamiento, en tratamiento inmunosupresor o que tengan EICH independientemente del tiempo desde el TPH.
- Receptores de trasplante de órgano sólido (menos de dos años o sometido a tratamiento inmunosupresor por sospecha de rechazo activo con independencia del tiempo desde el trasplante).
- Tratamiento sustitutivo renal (hemodiálisis y diálisis peritoneal).
- Inmunodeficiencias primarias: combinadas y de células B en las que se haya demostrado ausencia de respuesta vacunal.
- Tratamiento activo con quimioterapia mielotóxica para enfermedades oncológicas o hematológicas. Se excluye el uso de hormonoterapia, inhibidores de checkpoint inmunes u otros tratamientos que no condicionan aumento en el riesgo de infección (por ejemplo, anticuerpos monoclonales antidiana no mielotóxicos).
- Pacientes con tratamientos onco-hematológicos no citotóxicos con neutropenia (< 500 neutrófilos/mcL) o linfopenia (< 1000 linfocitos/mcL) en el momento de la infección.
- Infección por VIH con ≤200 cel/ml (analítica en los últimos 6 meses).
- Fibrosis quística.
- Síndrome de Down con 40 o más años de edad (nacidos en 1981 o antes).
- Tratamiento inmunosupresor con corticoides orales a altas dosis o durante tiempo prolongado y ciertos inmunomoduladores no biológicos:
- Tratamiento con corticoides orales a altas dosis de manera continuada (equivalente a ≥20 mg/día de prednisolona durante 10 o más días consecutivos en los treinta días previos).
- Tratamiento prolongado con corticoides orales a dosis moderadas (equivalente a ≥10 mg/día de prednisolona durante más de cuatro semanas consecutivas en los treinta días previos).
- Altas dosis de corticoides orales (equivalente a >40mg/día de prednisolona durante más de una semana) por cualquier motivo en los treinta días previos.
- Tratamiento en los tres meses anteriores con alguno de los siguientes fármacos inmunomoduladores no biológicos: metotrexato (>20 mg/semana o >15 mg/m2/sem, oral o subcutáneo), leflunomida, 6 mercaptopurina (>1,5 mg/kg/día) o azatioprina (>3 mg/kg/día), ciclosporina, micofenolato, tacrolimus (formas orales), sirolimus y everolimus en los tres meses previos.
- Tratamiento inmunosupresor con inmunomoduladores biológicos: Personas que han recibido en los tres meses anteriores (seis meses en caso de anti CD20) terapia específica con alguno de los fármacos de los siguientes grupos:
- Anticuerpos monoclonales anti CD20
- Inhibidores de la proliferación de células B (inmunomoduladores dirigidos a marcadores de las células B como CD40, CD19, CD38, CD79, Bcl6 entre otros)
- Proteínas de fusión supresoras de linfocitos T (inmunomoduladores dirigidos a proteínas de fusión que supriman la proliferación de los linfocitos T como el antígeno CD152 o CTLA4 entre otros)
- Inhibidores de la interleukina 1 (IL-1)
- Anticuerpos monoclonales anti-CD52
- Moduladores del receptor de la esfingosina-1-fosfato
- Inhibidores de la proteinquinasa.
- Inhibidores de la familia janus quinasa (JAK)
2. Personas no vacunadas* con >80 años.
3. Personas no vacunadas* con >65 años y con al menos un factor de riesgo para progresión**.
4. Personas vacunadas (> 6 meses) con > 65 años y con al menos un factor de riesgo para progresión**.
* Se consideran personas no vacunadas las persones que no han recibido la pauta de vacunación completa (incluidas las dosis de recuerdo) y no han padecido la enfermedad en los 3 últimos meses.
** Se consideran factores de riesgo de progresión:
- Enfermedad renal crónica: Pacientes con tasa de filtración glomerular inferior a 60 ml/min.
- Enfermedad hepática crónica: pacientes con una clasificación en la escala de Child-Pugh para gravedad de la enfermedad hepática de clase B o C (enfermedad hepática descompensada).
- Enfermedad neurológica crónica (Esclerosis múltiple, esclerosis lateral amiotrófica, miastenia gravis o enfermedad de Huntington).
- Enfermedades cardiovasculares, definidas como antecedentes de cualquiera de los siguientes: infarto de miocardio, accidente cerebrovascular (ACV), accidente isquémico transitorio (AIR), insuficiencia cardíaca, angina de pecho con nitroglicerina prescrita, injertos de revascularización coronaria, intervención coronaria percutánea, endarterectomía carotídea y derivación aórtica.
- Enfermedad pulmonar crónica (EPOC de alto riesgo (FEV1 postbroncodilatación < 50% o disnea (mMRC) de 2-4 o 2 o más exacerbaciones en el último año o 1 ingreso); asma con requerimiento de tratamiento diario).
- Diabetes con afectación de órgano diana.
- Obesidad (IMC≥35).
- Bajo peso (IMC≤18,5).

Condiciones de alto riesgo priorizadas en niños (hasta 18 años)
- Receptores de trasplante alogénico de progenitores hematopoyéticos o CAR-T, en los dos años tras el trasplante/tratamiento, en tratamiento inmunosupresor o que tengan EICH independientemente del tiempo desde el TPH.
- Inmunodeficiencias primarias: combinadas y de células B en las que se haya demostrado ausencia de respuesta vacunal.
- Tratamiento inmunosupresor con inmunomoduladores biológicos: Personas que han recibido en los tres meses anteriores (seis meses en caso de rituximab): fármacos anti CD20 o belimumab. Considerar en caso que el paciente haya estado en tratamiento con corticoides prolongados a dosis altas (> 2 mg/kg/dia 14 días o más, > 1 mg/Kg/día 28 días o más, > 20 mg/día en niños de más de 10 kg de peso 14 días o más).
- Pacientes que reciben quimioterapia de alta intensidad para tratamiento de leucemia, y aquellos con recuentos muy bajos de linfocitios (<100 cells/µL),
- Patología crónica compleja con dependencia tecnológica respiratoria.
- Receptores de trasplante de órgano sólido (menos de dos años) Solo si altamente inmunodeprimidos (eventos de rechazo, inducción) o comorbilidad (obesidad, no vacunados).
- Fibrosis quística (solo si afectación pulmonar grave).
- Considerar en adolescentes obesos IMC > 35 sin vacunar.
Diagrama de los criterios para valorar la administración de las nuevas alternativas terapéuticas antivirales frente a la infección por SARS-CoV-2
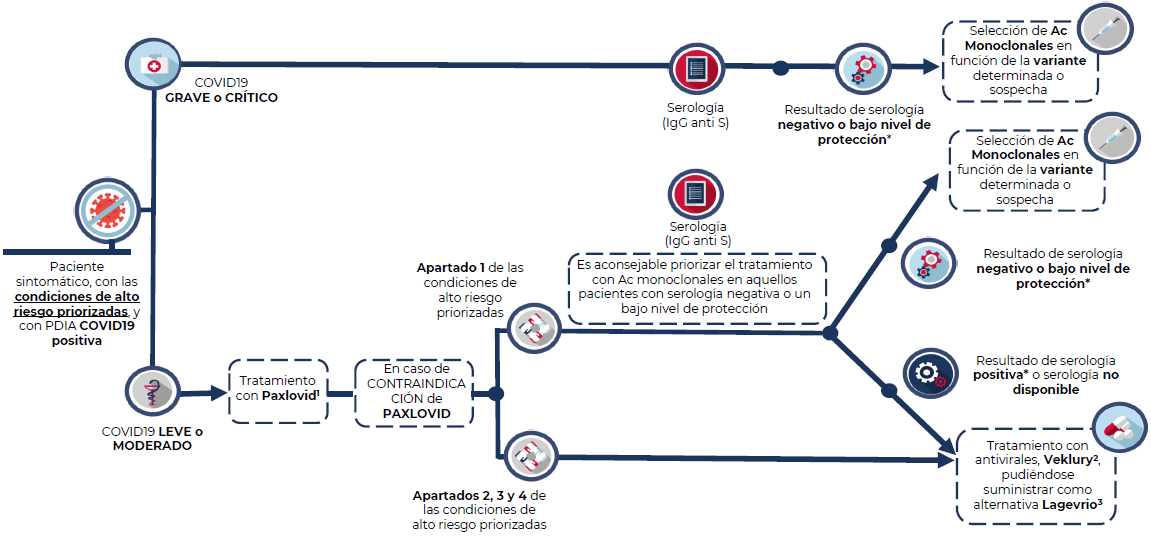
1. Paxlovid (nirmatrelvir/ritonavir): Se debe iniciar tratamiento dentro de los 5 días de evolución. 2. Veklury (remdesivir):.Se debe iniciar tratamiento dentro de los 7 días de evolución. 3. Lagevrio (molnupiravir): Se debe iniciar tratamiento dentro de los 5 días de evolución. * Bajo nivel de protección: Además de valorar la cuantificación de los títulos de anticuerpos frente a la proteína S, se debe tener en cuenta el grado de inmunosupresión del paciente. Es decir, la interpretación del resultado de la serología tendrá que llevarse a cabo junto con las características del paciente en cuanto a su grado de inmunosupresión y el riesgo individual de infección.
Anexo I
Tratamientos disponibles
Autorizados
Casirivimab e imdevimab son dos anticuerpos monoclonales recombinantes humanos que no tienen modificadas las regiones Fc. Casirivimab e imdevimab se unen a epítopos no superpuestos del dominio de unión al receptor de la proteína spike (RBD) del SARS-CoV-2. Esto evita que el RBD se una al receptor ACE2 humano, evitando así la entrada del virus en las células.
Ronapreve está indicado para:
- El tratamiento de enfermedad por COVID-19 en pacientes adultos y adolescentes de 12 años y mayores con un peso corporal de al menos 40 kg, que no requieran suplemento de oxígeno y que tengan mayor riesgo de progresar a COVID-19 grave.
- La prevención de COVID-19 en pacientes adultos y adolescentes de 12 años y mayores con un peso corporal de al menos 40 kg.
La eficacia y la seguridad de Ronapreve se han estudiado en distintos ensayos clínicos de fase III en pacientes no hospitalizados y hospitalizados por COVID-19, y como terapia preventiva. En la actualidad, Ronapreve no está autorizado en pacientes hospitalizados. El uso en esta indicación se apoya en los resultados del ensayo RECOVERY.
El ensayo RECOVERY (Randomised Evaluation of COVID-19 thERapY) es un ensayo promovido por el Sistema de Salud de Reino Unido con el objetivo de evaluar los potenciales tratamientos frente a la COVID-19. En este estudio se ha evaluado la eficacia y seguridad de Ronapreve en adultos hospitalizados. Se aleatorizaron 9785 pacientes hospitalizados con COVID-19 para recibir la atención habitual más el tratamiento combinado de anticuerpos (casirivimab 4g con imdevimab 4g por infusión intravenosa) o la atención habitual sola. De estos, aproximadamente un tercio eran seronegativos al inicio del estudio (es decir, no habían desarrollado una respuesta de anticuerpos natural propia.
Entre los pacientes que eran seronegativos al inicio del estudio, la combinación de anticuerpos redujo significativamente el resultado primario de mortalidad a 28 días en una quinta parte en comparación con la atención habitual sola (el 24% de los pacientes en el grupo de combinación de anticuerpos murió vs 30% de los pacientes en el grupo de atención habitual; RR= 0,80; IC 95% 0,70-0,91; p = 0,001).
Los resultados del estudio RECOVERY se pueden consultar en el siguiente link:
https://www.recoverytrial.net/
Datos in vitro actualizados, muestran que Ronapreve tiene una actividad reducida frente a la variante ómicron. La decisión sobre el uso de Ronapreve debe tener en cuenta lo que se conoce sobre las características de los virus del SARS-CoV-2 circulantes y la información disponible sobre los patrones de sensibilidad a Ronapreve.
La Agencia recomienda a los profesionales sanitarios que consulten la ficha técnica de este medicamento:
https://cima.aemps.es/cima/dochtml/ft/1211601002/FT_1211601002.html https://cima.aemps.es/cima/dochtml/ft/1211601001/FT_1211601001.html
Acceso
El acceso deberá realizarse a través de los cauces habituales, no siendo necesaria su solicitud a través de medicamentos en situaciones especiales excepto en aquellas CC.AA. que no cuenten con un depósito estratégico. En estos casos, el acceso a este medicamento se hará de forma individualizada a través de la aplicación de medicamentos en situaciones especiales. En la aplicación se encuentra toda la información necesaria para solicitarlo.
Sotrovimab es un anticuerpo monoclonal (mAb) IgG1 humanizado que se une a un epítopo altamente conservado en el dominio de unión al receptor (RBD) de la proteína spike del SARS-CoV-2.
Sotrovimab está indicado en el tratamiento de la COVID19 en adultos y adolescentes que no requieren oxígeno suplementario y que tienen un mayor riesgo de progresar a estadios más graves de COVID-19.
La eficacia y seguridad de sotrovimab han sido evaluadas para la indicación autorizada mediante un estudio pivotal de fase III COMET-ICE, en adultos no vacunados con menos de 5 días de síntomas, que muestra que el tratamiento intravenoso con sotrovimab resultó en una reducción del 79% (reducción del riesgo relativo ajustado) (p <0,001) en las hospitalizaciones por cualquier causa durante más de 24 horas o muerte por cualquier causa el día 29 en comparación con placebo, cumpliendo así con el criterio de valoración principal del ensayo. En números absolutos, 30 (6%) de los 529 voluntarios del grupo de placebo progresaron, en comparación con seis (1%) de los 528 pacientes que recibieron sotrovimab. En los ensayos clínicos realizados hasta la fecha, sotrovimab ha sido bien tolerado. Las reacciones adversas más frecuentes son la hipersensibilidad y las reacciones relacionadas con la infusión, que se observan en aproximadamente el 2% y el 1% de los casos, respectivamente.
En pacientes hospitalizados por COVID-19, el ensayo clínico TICO, que evalúa la eficacia y seguridad de sotrovimab 500 mg o la combinación de BRII-196 1000 mg junto a BRII-198 1.000 mg frente a placebo, se suspendió tras un análisis intermedio de futilidad en los primeros 546 pacientes reclutados, tras no observarse beneficio clínico a los 5 días del tratamiento. En el ensayo clínico RECOVERY se está evaluado actualmente la eficacia y seguridad de sotrovimab 1.000 mg en pacientes hospitalizados por COVID-19.
Datos in vitro actualizados, muestran que sotrovimab tiene una actividad reducida frente a la variante BA.2.
La evidencia del uso de sotrovimab en paciente con inmunosupresión de alto riesgo es muy limitada y se trata de un uso fuera de indicación.
La Agencia recomienda a los profesionales sanitarios que consulten la ficha técnica de este medicamento:
https://cima.aemps.es/cima/dochtml/ft/1211562001/FT_1211562001.html
Acceso
El acceso deberá realizarse a través de los cauces habituales, no siendo necesaria su solicitud a través de medicamentos en situaciones especiales excepto en aquellas CC.AA. que no cuenten con un depósito estratégico, y las CCAA que lo indiquen expresamente. En estos casos, el acceso a este medicamento se hará de forma individualizada a través de la aplicación de medicamentos en situaciones especiales. En la aplicación se encuentra toda la información necesaria para solicitarlo.
Tixagevimab y cilgavimab son dos anticuerpos monoclonales IgG1 humanos recombinantes. Tixagevimab y cilgavimab pueden unirse simultáneamente a regiones no superpuestas del dominio de unión al receptor de la proteína de la espícula (Spike Protein Receptor Binding Domain, RBD) de SARS-CoV-2.
Evusheld está indicado como profilaxis previa a la exposición de COVID-19 en adultos y adolescentes a partir de 12 años de edad que pesen al menos 40 kg y para el tratamiento de adultos y adolescentes (a partir de 12 años de edad que pesen al menos 40 kg) con COVID-19, que no requieran oxígeno suplementario y que tengan un riesgo alto de progresar a COVID-19 grave.
La eficacia y seguridad de Evusheld para el tratamiento han sido evaluadas para la indicación autorizada mediante un estudio pivotal de fase III TACKLE, en adultos no vacunados con menos de 7 días de síntomas, que muestra que el tratamiento intramuscular con Evusheld resultó en una reducción del 67% (reducción del riesgo relativo) (IC 95% de 31, 84) en pacientes no hospitalizados dosificados en los primeros 5 días desde el inicio de los síntomas (p= 0,002).
La Agencia recomienda a los profesionales sanitarios que consulten la ficha técnica de este medicamento:
https://cima.aemps.es/cima/pdfs/ft/1221651001/FT_1221651001.pdf
Acceso
Los envases de Evusheld fueron distribuidos en función de la población a las CC.AA. que indicaron los centros a los cuales se les asignaban las dosis, la solicitud de estos envases se realiza a través de la aplicación de medicamentos en situaciones especiales (MSE). Los envases pueden ser utilizados para el tratamiento siempre que se cumplan los criterios establecidos en este documento.
Es un antiviral análogo de nucleótido, que interfiere con la polimerización del ARN del virus.
Veklury está autorizado para el tratamiento de la enfermedad por coronavirus 2019 (COVID-19) en:
- Adultos y adolescentes (de 12 a menos de 18 años de edad y que pesen al menos 40 kg) con neumonía que requieren oxígeno suplementario (oxígeno de alto o bajo flujo u otra ventilación no invasiva al inicio del tratamiento).
- Adultos que no requieren oxígeno suplementario y que presentan un riesgo más alto de evolucionar a COVID-19 grave.
La indicación reflejada en el segundo punto, se encuentra pendiente de decisión de financiación.
La eficacia y seguridad de remdesivir en adultos que no requieren oxígeno suplementario y que presentan un riesgo más alto de evolucionar a COVID-19 grave han sido evaluadas mediante un estudio pivotal de fase III aleatorizado, doble ciego, controlado con placebo y multicéntrico para evaluar el tratamiento con remdesivir en un entorno ambulatorio en 562 pacientes adultos con COVID-19 confirmado, no vacunados y al menos un factor de riesgo de mala evolución. Los pacientes tratados con remdesivir recibieron 200 mg el día 1 y 100 mg una vez al día en los días posteriores durante un total de 3 días de tratamiento administrado por vía intravenosa.
La variable primaria fue la proporción de pacientes con hospitalización relacionada con COVID-19 (definida como al menos 24 horas de cuidados intensivos) o mortalidad por todas las causas a los 28 días. En un análisis de 562 pacientes remdesivir mostró una reducción estadísticamente significativa del 87% (0,7% [2/279]) en comparación con el placebo (5,3% [15/283]) p=0,008. No se observaron muertes el día 28.
La Agencia recomienda a los profesionales sanitarios que consulten la ficha técnica de este medicamento:
https://cima.aemps.es/cima/dochtml/ft/1201459002/FT_1201459002.html
Acceso
El acceso deberá realizarse a través de los cauces habituales, no siendo necesaria su solicitud a través de medicamentos en situaciones especiales. El uso de este medicamento debe realizarse de acuerdo a los protocolos de manejo clínico de cada hospital.
Paxlovid contiene dos fármacos: nirmatrelvir, un inhibidor de la proteasa principal del SARS-CoV2 y ritonavir, un inhibidor del citocromo CYP3A4, un potenciador farmacocinético de nirmatrelvir, inhibiendo su metabolismo. Ritonavir produce múltiples interacciones que es necesario validar antes del uso de Paxlovid.
Paxlovid está indicado para el tratamiento de la enfermedad por coronavirus 2019 (COVID-19) en adultos que no requieren aporte de oxígeno suplementario y que tienen un riesgo alto de progresar a COVID-19 grave.
La eficacia de Paxlovid se basa en el análisis intermedio y en el análisis final de EPIC-HR (Evaluación de la Inhibición de la Proteasa para el COVID-19 en pacientes de Alto Riesgo, por sus siglas en inglés), un estudio en fase 2/3, aleatorizado, doble ciego y controlado con placebo en participantes adultos sintomáticos no hospitalizados con un diagnóstico confirmado por laboratorio de infección por SARS-CoV-2. Todos los pacientes no estaban vacunados y no debían haberse infectado previamente por SARS-CoV-2. Compara el uso de Paxlovid administrado oralmente dos veces al día durante 5 días en pacientes adultos sintomáticos no hospitalizados con COVID-19 confirmado, leve-moderado y con un factor de riesgo preespecificado para progresión a enfermedad grave o pacientes de 60 o mayores independientemente de la condición médica crónica preespecificada.
La variable principal compuesta fue hospitalización relacionada con COVID-19 o muerte por cualquier causa durante los 28 días de seguimiento. El análisis preliminar mostró una reducción relativa del 89 % en el riesgo de hospitalización o muerte por cualquier causa relacionada con la COVID-19 en comparación con el placebo en pacientes tratados dentro de los tres días posteriores al inicio de los síntomas (variable principal).
El 0,8 % de los pacientes que recibieron Paxlovid fueron hospitalizados hasta el día 28 después de la aleatorización (3/389 hospitalizados sin muertes), en comparación con el 7,0 % de los pacientes que recibieron placebo y fueron hospitalizados o fallecieron (27/385 hospitalizados con 7 muertes posteriores).
Se observaron reducciones similares en la hospitalización o muerte relacionadas con COVID-19 en el análisis final en pacientes tratados dentro de los cinco días posteriores al inicio de los síntomas.; El 0,8 % de los pacientes que recibieron Paxlovid fueron hospitalizados hasta el día 28 después de la aleatorización (8/1.039 hospitalizados, sin muertes), en comparación con el 6,3 % de los pacientes que recibieron un placebo (66/1.046 hospitalizados con 12 muertes posteriores). Esto hace que sea necesario tratar a 18 personas en sus primeros 5 días de síntomas para evitar 1 ingreso hospitalario.
A pesar de excluir en el ensayo tanto a pacientes vacunados como a pacientes que habían pasado la enfermedad, aproximadamente la mitad de los pacientes incluidos (1.068/2.085) tenía serología positiva para el SARS-CoV2 y la eficacia en estos pacientes fue unas 10 veces menor que en aquellos con serología negativa (eficacia absoluta del 1,34% y del 10,25% respectivamente). En pacientes seronegativos, el 1,4% de los pacientes que recibieron Paxlovid fueron hospitalizados (7/487) en comparación con el 11,5% de los que recibieron el placebo (58/505) y en pacientes seropositivos, el 0,2% de los pacientes que recibieron Paxlovid fueron hospitalizados (1/540) en comparación con el 1,5% de los que recibieron el placebo (8/528). En el caso de pacientes seronegativos sería necesario tratar a 10 pacientes para evitar un ingreso hospitalario, mientras que en pacientes seropositivos, sería necesario tratar a 75 pacientes para evitar un ingreso.
La Agencia recomienda a los profesionales sanitarios que consulten la ficha técnica de este medicamento:
https://cima.aemps.es/cima/pdfs/ft/1221625001/FT_1221625001.pdf
Acceso
El acceso deberá realizarse a través de los cauces habituales, no siendo necesaria su solicitud a través de medicamentos en situaciones especiales. Sin embargo, debido al perfil de interacciones y advertencias especiales de uso, se requiere una validación farmacéutica previa a su dispensación. Este circuito de validación lo establecerá cada CC.AA. y debe ser inferior a 24h, salvo en el caso de pacientes privados y pertenecientes a MUFACE, MUGEJU e ISFAS con asistencia sanitaria privada, cuyo acceso se tramita a través de MSE.
No autorizados que cuentan con recomendación de uso del Comité de Medicamentos de Uso Humano
El comité de medicamentos de uso humano (por sus siglas en inglés CHMP) de la EMA ha emitido recomendaciones sobre el uso de Lagevrio (molnupiravir) para el tratamiento de COVID-19. El medicamento, que actualmente no está autorizado en la UE, se puede usar para tratar a adultos con COVID-19 que no requieren oxígeno suplementario y que tienen un mayor riesgo de desarrollar COVID-19 grave. Molnupiravir debe administrarse lo antes posible después del diagnóstico de COVID-19 y dentro de los 5 días posteriores al inicio de los síntomas. El medicamento, que está disponible en cápsulas, debe tomarse dos veces al día durante 5 días.
La EMA emitió esta recomendación para ayudar a las autoridades nacionales que pueden decidir sobre un posible uso temprano del medicamento antes de la autorización de comercialización, por ejemplo, en entornos de uso de emergencia, a la luz del aumento de las tasas de infección y muerte por COVID-19 en la UE.
La recomendación sigue a una revisión de los datos, incluidos los datos sobre la calidad del medicamento y los resultados de los estudios completados y en curso. Como parte de este consejo, se evaluaron los resultados provisionales del estudio principal en pacientes no hospitalizados y no vacunados con al menos una afección subyacente que los pone en riesgo de COVID-19 grave. Molnupiravir, cuando se administró en una dosis de 800 mg dos veces al día, redujo el riesgo de hospitalización y muerte cuando el tratamiento se inició dentro de los 5 días posteriores al inicio de los síntomas. Aproximadamente un mes después del inicio del tratamiento, el 7,3% de los pacientes (28 de 385) que tomaron molnupiravir en comparación con el 14,1% (53 de 377) de los pacientes que tomaron placebo habían sido hospitalizados o habían fallecido; ninguno de los pacientes del grupo de molnupiravir murió en comparación con ocho pacientes del grupo de placebo.
Aunque la EMA no ha evaluado todavía los datos finales del estudio, ya están disponibles los datos de todos los participantes incluidos en el mismo (n = 1433). En esta población, el 6,8% de los pacientes (48 de 709) que tomaron molnupiravir en comparación con el 9,7% (68 de 699) de los pacientes que tomaron placebo habían sido hospitalizados o habían fallecido, lo que supone una reducción del riesgo absoluto del 3,0% (IC95% 0,1-5,9; p= 0,0218) y una reducción del riesgo relativo del 30%. Se informaron nueve muertes en el grupo de placebo y una en el grupo de molnupiravir.
Esta recomendación puede consultarse en el siguiente link:
https://www.ema.europa.eu/en/news/ema-issues-advice-use-lagevrio-molnupiravir-treatment-covid-19
Los datos finales del estudio publicados por el laboratorio pueden consultarse en el siguiente link:
https://www.merck.com/news/merck-and-ridgeback-biotherapeutics-provide-update-on-results-from-move-out-study-of-molnupiravir-an-investigational-oral-antiviral-medicine-in-at-risk-adults-with-mild-to-moderate-covid-19/
Acceso
En paciente de alto riesgo y enfermedad leve-moderada no hospitalizado o en el contexto de un brote nosocomial, se priorizará el uso de remdesivir por ser una alternativa autorizada y comercializada. En caso de considerar su uso, el acceso a este medicamento se hará de forma individualizada a través de la aplicación de medicamentos en situaciones especiales. En la aplicación de medicamentos en situación especial se encuentra toda la información necesaria para solicitarlo.
Anexo II
Grupo de Trabajo Técnico
JOSE RAMÓN ARRIBAS LÓPEZ
Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC)
CRISTINA CALVO REY
Asociación Española de Pediatría (AEP)
RAFAEL DE LA CÁMARA LLANZA
Sociedad Española de Hematología y Hemoterapia (SEHH)
OLGA DELGADO SANCHEZ
Sociedad Española de Farmacia Hospitalaria (SEFH)
JULIO GARCÍA RODRÍGUEZ
Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC)
JUAN GONZALEZ DEL CASTILLO
Sociedad Española de Medicina de Urgencias y Emergencias (SEMES)
ANTONIO LALUEZA BLANCO
Sociedad Española de Medicina Interna (SEMI)
MARCOS LÓPEZ HOYOS
Sociedad Española de Inmunología (SEI)
ROSARIO MENÉNDEZ VILLANUEVA
Sociedad Española de Neumología y Cirugía Torácica (SEPAR)
ARANTXA SANCHO LÓPEZ
Sociedad Española de Farmacología Clínica (SEFC)
EMILIO ALEGRE DEL REY
CC.AA. Andalucía
AINHOA ARANGUREN OYARZÁBAL
CC.AA. Madrid
TOMÁS CARO-PATÓN CARMONA
CC.AA. Castilla y León
Mª DOLORES FRAGA FUENTES
D.G. de Cartera Común de Servicio del SNS y Farmacia
ANTONI VALLANO FERRAZ
CC.AA. Cataluña
MILENA PERAITA EZCURRA
Agencia Española de Medicamentos y Productos Sanitarios (AEMPS)
ANTONIO LÓPEZ NAVAS
Agencia Española de Medicamentos y Productos Sanitarios (AEMPS)
1National Institute for Biological Standards and Control. First WHO International Reference Panel for anti-SARS-CoV-2 immunoglobulin. NIBSC code: 20/268. Instructions for use (version 3·0). Potters Bar: National Institute for Biological Standards and Control, 2020.
2Nature Medicine volume 27, pages2032–2040 (2021)
3Gilbert et al., Science 375, 43–50 (2022)
4Goldblatt et al. Vaccine 40 (2022) 306–315