

Desde 2003 existe en la UE un procedimiento de autorización específico para los medicamentos biosimilares mediante el cual se valoran los estudios realizados con el medicamento biosimilar en comparación con el medicamento de referencia. Como la mayoría de medicamentos biosimilares autorizados son biotecnológicos, su autorización sigue el denominado procedimiento centralizado, coordinado por la EMA (procedimiento obligado para la evaluación y autorización de medicamentos biotecnológicos en la UE).
El período de evaluación tiene una duración de 210 días y concluye con la opinión científica del Comité de Medicamentos para Uso Humano (CHMP, por sus siglas en inglés) de la EMA en el que están representadas todas las agencias reguladoras de los Estados miembros; en España, la AEMPS.
Una vez autorizado, el precio y financiación del medicamento se deciden a nivel nacional, en España es competencia de la Dirección General de Cartera Común de Servicios del SNS y Farmacia. El primer medicamento biosimilar se autorizó en la UE en 2006 y actualmente se ha convertido en la región que más biosimilares tiene autorizados.
Los biosimilares sólo pueden comercializarse una vez que ha expirado el periodo de exclusividad de los datos del medicamento biológico «de referencia». En general, esto significa que el medicamento biológico de referencia debe haber estado autorizado durante al menos ocho años antes de que otra empresa pueda comercializar un medicamento biológico similar.
Evaluación de equivalencia de medicamentos biosimilares
El procedimiento para establecer la equivalencia entre ambos debe ser un proceso secuencial, es decir, comenzando con la evaluación de la estructura molecular y actividad farmacológica del principio activo. Una vez demostrado que ambas moléculas son comparables tanto estructural como funcionalmente, se evaluará el tipo y el alcance de los estudios no clínicos y clínicos.
Para la evaluación de esta equivalencia es necesario presentar datos obtenidos por diferentes tipos de estudios comparados.
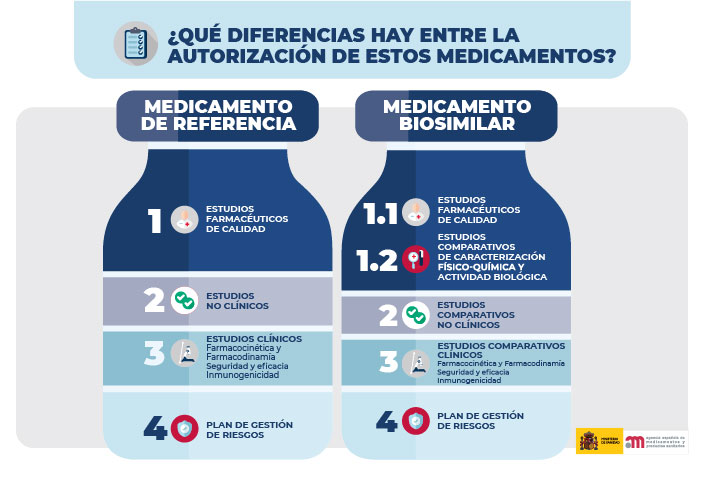
Los estudios in vitro comparan la estructura proteica y la función biológica utilizando técnicas sensibles capaces de detectar diferencias mínimas con relevancia clínica entre el biosimilar y su medicamento de referencia. Estos estudios son mucho más sensibles que los ensayos clínicos para la detección de dichas diferencias, ya que suele existir variabilidad entre los sujetos humanos que participan en los ensayos. Si se detectan diferencias que puedan afectar a la seguridad clínica, a la eficacia o a la inmunogenicidad, sería necesario realizar más estudios (por ejemplo, estudios comparativos clínicos o preclínicos, pasos 2 y 3).
Estos estudios incluyen estudios farmacodinámicos in vitro que analizan la unión y la activación (o inhibición) de las dianas fisiológicas y los efectos fisiológicos inmediatos en las células. Solo se efectúan estudios farmacodinámicos in vivo (modelos animales) si no existe un modelo adecuado in vitro. Únicamente se requieren estudios toxicológicos in vivo en determinados casos, por ejemplo, cuando el biosimilar es producido en un nuevo tipo de célula u organismo, o cuando la formulación incluye nuevos excipientes que no se han utilizado previamente.
El objetivo de los estudios en humanos no es demostrar la seguridad y la eficacia en los pacientes, ya que estos parámetros han sido establecidos para el medicamento de referencia. Los ensayos clínicos están diseñados para confirmar la biosimilitud y para abordar cualquier cuestión que quedase pendiente de los estudios analíticos o funcionales realizados previamente
Es importante resaltar que la evaluación de los biosimilares se lleva a cabo por los mismos grupos de expertos de las agencias reguladoras, en España los técnicos de la AEMPS, y con las mismas exigencias que las de los innovadores. Así, cuando se aprueba un biosimilar, se garantiza que:
- Cumple todos los requisitos de calidad que se exige a un innovador
- Las diferencias con respecto al medicamento de referencia son, en general, parecidas a las que podrían existir entre diferentes lotes del innovador
La regulación permite el desarrollo de biosimilares de cualquier tipo de medicamento biológico, pero en la práctica esto está limitado a aquellos que pueden ser analizados de forma exhaustiva a nivel físico-químico y funcional.
Además, se exige que el medicamento de referencia esté comercializado en el Espacio Económico Europeo (EEE, es decir, la UE más los estados asociados de Noruega, Islandia y Liechtenstein), y que los principales estudios de biosimilitud han de hacerse con lotes adquiridos en el mercado europeo. Se permite el uso de lotes de otros mercados como apoyo y siempre que hayan sido autorizados por autoridades reguladoras con requisitos tan estrictos como los de los países del EEE.
Por tanto, los biosimilares aprobados en la UE son intercambiables desde un punto de vista científico, lo que significa que un biosimilar puede utilizarse en lugar de su producto de referencia, o viceversa.
Asimismo, un biosimilar puede utilizarse en lugar de otro biosimilar del mismo producto de referencia. Los Estados miembros de la UE siguen decidiendo qué medicamentos biológicos pueden recetarse en su país y si se permite la sustitución automática o no por el biosimilar equivalente.