 Recomendaciones y aclaraciones para la aplicación de los Dispositivos de Seguridad de Medicamentos de uso humano
Recomendaciones y aclaraciones para la aplicación de los Dispositivos de Seguridad de Medicamentos de uso humano
- Compartir:
-

-

-

-

Última actualización: 27/8/2019
NOTA: este documento de preguntas y respuestas debe considerarse en el contexto regulatorio del momento en el cual se publica, sin perjuicio de la legislación vigente ni de los avances en los requerimientos regulatorios que puedan tener lugar con posterioridad. En este sentido, este documento se emite sin perjuicio del obligado cumplimiento del REGLAMENTO DELEGADO (UE) 2016/161 DE LA COMISIÓN de 2 de octubre de 2015 que completa la Directiva 2001/83/CE del Parlamento Europeo y del Consejo estableciendo disposiciones detalladas relativas a los dispositivos de seguridad que figuran en el envase de los medicamentos de uso humano, de la última versión del Documento de Preguntas y Respuestas (Q&A) en relación con los Dispositivos de Seguridad de Medicamentos de uso humano publicado por la Comisión Europea y sus modificaciones futuras y de la Nota Informativa sobre Implementación de los Dispositivos de Seguridad en las Autorizaciones de Comercialización de los medicamentos de uso humano del 12 de Julio de 2017, en adelante “NI DS”, publicada por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS).
ÍNDICE DE CONTENIDOS
- INTRODUCCIÓN
-
ASPECTOS RELACIONADOS CON EL ETIQUETADO Y PROCEDIMIENTOS DE REGISTRO
- ¿CÓMO EXPRESAR LA INFORMACIÓN DEL IDENTIFICADOR ÚNICO (IU)?
- ¿CÓMO EXPRESAR EL LOTE Y CADUCIDAD EN EL EMBALAJE EXTERIOR?
- MAQUETAS DE LOS FORMATOS NO COMERCIALIZADOS
- ¿CÓMO SE INCORPORARÁ A LOS DATOS DEL REGISTRO EL GTIN DEL FORMATO CUANDO EL TAC OPTE POR ESTA ALTERNATIVA PARA IDENTIFICAR EL PC?
- OFICIADO DE NOTIFICACIONES CONFORME AL ARTÍCULO 61(3)
- EN RELACIÓN A LAS TASAS CUANDO SE TRATA DE UN MEDICAMENTO AUTORIZADO POR RM/DC
- CODIFICACIÓN DE LOS ELEMENTOS DEL IDENTIFICADOR ÚNICO
- NECESIDAD DE INCLUIR EL BRAILLE EN 2 Ó 3 LÍNEAS Y ABREVIATURAS
- EN EL CASO DE MEDICAMENTOS QUE NO ESTUVIERAN AFECTADOS POR LA OBLIGACIÓN DE INCORPORAR DISPOSITIVOS DE SEGURIDAD, ¿DEBE ACTUALIZARSE LA PLANTILLA QRD INCLUYENDO LAS SECCIONES 17 Y 18 ANTES DEL 9 DE FEBRERO DE 2019?
- EN EL CASO DE MEDICAMENTOS QUE NO ESTUVIERAN AFECTADOS POR LA OBLIGACIÓN DE INCORPORAR DISPOSITIVOS DE SEGURIDAD PERO INCORPOREN UN CÓDIGO DE BARRAS BIDIMENSIONAL PARA OTROS OBJETIVOS DIFERENTES (P. EJ. PRECIO, REEMBOLSO, LOTE, CADUCIDAD…), ¿CÓMO DEBE NOTIFICARLO A LA AEMPS?
- OTRAS CONSIDERACIONES A TENER EN CUENTA
- DISPOSITIVOS CONTRA MANIPULACIONES (DCM)
-
FABRICACIÓN Y CONTROL
- LA INSTALACIÓN DE EQUIPOS PARA LA SERIALIZACIÓN DE LOS ENVASES, ¿REQUIERE ALGÚN TIPO DE COMUNICACIÓN A LA AEMPS?
- ETIQUETAS
- MUESTRAS GRATUITAS
- ¿CUÁLES SON LAS CONDICIONES QUE SE DEBEN CUMPLIR EN CASO DE QUE UN LABORATORIO ENCARGUE LA IMPRESIÓN DEL IU A UN TERCERO CONTRATADO?
- EN EL CASO DE UTILIZAR ENVASES PRE-SERIALIZADOS, ¿ES POSIBLE CARGAR EN EL REPOSITORIO TODOS LOS IU RECIBIDOS DEL PROVEEDOR QUE HA PRE-SERIALIZADO LOS ENVASES?
- DISPOSITIVO CONTRA MANIPULACIONES (DCM)
- IMPORTACIONES PARALELAS
- OPERACIONES DE LOS DISPOSITIVOS DE SEGURIDAD
- MEDIDAS PARA DISMINUIR EL NÚMERO DE ALERTAS
1. INTRODUCCIÓN (Índice)
El presente documento está dirigido a la Industria Farmacéutica y pretende responder a las preguntas frecuentes en relación con la aplicación de la NI DS y recoge la respuesta que desde la AEMPS se da a las preguntas surgidas desde la publicación de la mencionada NI.
La información contenida en este documento no es exhaustiva y se considera responsabilidad del Titular de la Autorización de Comercialización (TAC) asegurar el cumplimiento de los requerimientos establecidos en la normativa más arriba referenciada.
La versión que aquí se presenta es el resultado del trabajo de técnicos del Departamento de Medicamentos de Uso Humano y Departamento de Inspección y Control de Medicamentos de la AEMPS, y se ha consultado con profesionales que trabajan en compañías farmacéuticas, por lo que será una herramienta útil para el sector. No obstante, se trata de un documento dinámico, que podrá estar sujeto a cambios en función de las necesidades, nuevas preguntas que vayan surgiendo, así como a la publicación de nuevas versiones del Documento de Preguntas y Respuestas de la Comisión Europea. Por tanto, su carácter es informativo y podrá ser modificado.
Cualquier consulta, aclaración o propuesta de mejora podrá dirigirse a la dirección: dgestion@aemps.es
2. ASPECTOS RELACIONADOS CON EL ETIQUETADO Y PROCEDIMIENTOS DE REGISTRO (Índice)
2.1 ¿CÓMO EXPRESAR LA INFORMACIÓN DEL IDENTIFICADOR ÚNICO (IU)? (Índice)
Se muestran ejemplos consultados (el Datamatrix y los números empleados son ficticios):
PC: código del producto
SN: número de serie
NN: número nacional de reembolso
Lote/Lot: Lote
CAD/EXP: Caducidad
| Identificador único: Información en caracteres visuales |
Caso | Permiso | ||
|---|---|---|---|---|

|
PC SN Lote CAD |
08470008722513 6874352687 L201JX32 Ene 2021 |
CASO 1: La información adyacente al Datamatrix incluye PC, SN, lote y caducidad. | PERMITIDO |
|
|
PC SN |
08470008722513 6874352687 |
CASO 2: La información sobre lote y caducidad incluye en otra zona del embalaje exterior. | PERMITIDO |
|
|
PC SN Lote CAD |
08470008722513 6874352687 L201JX32 Ene 2021 |
CASO 3: PC está pre-impreso. La información adyacente al Datamatrix incluye PC, SN, lote y caducidad. | PERMITIDO |
|
|
PC SN |
08470008722513 6874352687 |
CASO 4: PC está pre-impreso. La información sobre lote y caducidad se incluye en otra zona del embalaje exterior. | PERMITIDO |
|
|
PC SN Lote CAD |
08470008722513 6874352687 L201JX32 Ene 2021 |
CASO 5: La información está toda pre-impresa en vez de ser impresa en línea. | PERMITIDO |
|
|
PC SN |
08470008722513 6874352687 |
CASO 6: La información de PC y SN está pre-impresa. La información sobre lote y caducidad se incluye en otra zona del embalaje exterior. | PERMITIDO |
|
|
PC SN Lote CAD |
08470008722513 6874352687 L201JX32 Ene 2021 |
CASO 7: La información sobre PC y SN está pre-impresa. Lote y caducidad se imprimen en línea en la zona adyacente. | PERMITIDO |
|
|
PC SN |
(01) 8470008722513 (21) 6874352687 |
CASO 8: Se incluyen los prefijos de los elementos del IU en caracteres visuales. | PERMITIDO1 |
|
|
PC SN Lote CAD |
(01) 08470008722513 (21) 6874352687 (10) L201JX32 (17) Ene 2021 |
CASO 9: Se incluyen los prefijos (01), (21), de los elementos del IU así como los prefijos (10), (17) para Lote y CAD en caracteres visuales. | PERMITIDO1 |
|
|
PC SN Lote CAD |
(01) 08473543123678 (21) 687435268 (10) L201JX32 Ene 2021 |
CASO 10: Se suprime el (17) en caducidad para evitar toda confusión con el año. | PERMITIDO1 |
|
|
PC SN Lote CAD |
(01NTIN) 08470008722513 (21) 6874352687 (10) L201JX32 (17) Ene 2021 |
CASO 11: Se incluyen las siglas NTIN o GTIN y los prefijos (01), (21), (10), (17) de los elementos del IU en forma legible y otra información adicional para el PC. | NO PERMITIDO1 |
|
|
PC SN NN Lote CAD |
08470008722513 6874352687 872251.3 L201JX32 Ene 2021 |
CASO 12: Se incluye el NN como información adicional, con un PC NTIN, duplicando el Número Nacional de reembolso (que debe figurar en el ángulo superior derecho de la cara principal o de la blue box). | PERMITIDO |
|
|
PC SN NN Lote CAD |
08473543123678 6874352687 872251.3 L201JX32 Ene 2021 |
CASO 13: Se incluye el NN como información adicional, con un PC GTIN, duplicando el Número Nacional de reembolso (que debe figurar en el ángulo superior derecho de la cara principal o de la blue box). | PERMITIDO |
|
|
PC: SN: Lote: CAD: |
08473543123678 687435268 L201JX32 Ene 2021 |
CASO 14: Los prefijos PC y SN se indican como en la plantilla del QRD con dos puntos (:). También se permiten (:) en Lote y Caducidad. | PERMITIDO |
|
|
PC Lote CAD SN |
08473543123678 L201JX32 Ene 2021 687435268 |
CASO 15: La información se mantiene en otro orden. | PERMITIDO2 |
|
|
PC/SN/LOT/EXP: 08470006723459 98876582837645683 Bhj7869d 02-2020 |
CASO 16: Disposición en línea de los títulos de los elementos del IU: (PC/SN/LOT/EXP) se indican a modo de título pre-impreso. | PERMITIDO3 | |
|
|
PC 08470006723459 LOT/EXP/SN Bhj7869d 02-2020 98876582837645683 |
CASO 17: Disposición en línea de los títulos de los elementos del IU (LOT/EXP/SN) | PERMITIDO3 | |
|
|
PC SN Lote CAD |
-- N.A. L201JX32 Ene 2021 |
CASO 18: Durante el período de transición (hasta el 09 febrero 2019), en las líneas del identificador único que corresponden a PC y/o SN figura impreso “NA” o “-“ | NO PERMITIDO |
|
|
PC SN Lote CAD |
L201JX32 Ene 2021 |
CASO 19: Durante el período de transición (hasta el 09 febrero 2019), las líneas del identificador único que corresponden a PC y/o SN están vacías. | PERMITIDO |
|
|
CAD/Lote
PC SN |
10.2020 3X6T 08470006540206 3BSBGH62B2 |
CASO 20: poner pre-impreso el texto “CAD/Lote” y debajo el resto de la información, identificando sólo las líneas de PC y SN. | PERMITIDO |
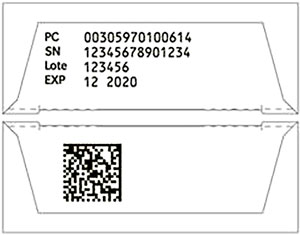
|
CASO 21: Cuando las dimensiones del envase no permitan que aparezcan en la misma cara Datamatrix e información en caracteres visuales, ¿está permitido que aparezcan en dos caras diferentes? | PERMITIDO4 | ||
|
|
GTIN SN Lote CAD |
08473543123678 687435268 L201JX32 Ene 2021 |
CASO 22: poner GTIN en vez de PC. | NO PERMITIDO |
|
|
PC (GTIN) SN |
CASO 23: Multilingüe. | CONTACTAR CON AEMPS PARA RESOLVER CASO POR CASO | |
|
PC SN Lote CAD |
L201JX32 Ene 2021 |
CASO 24: Durante el período de transición (hasta el 09 febrero 2019), se permite sin Datamatrix y PC y SN pre-impreso vacío. Lote y Caducidad ya impresos previamente. | PERMITIDO | |
|
PC SN Lote CAD |
08470008722513
L201JX32 Ene 2021 |
CASO 25: PC está pre-impreso, durante el período de transición (hasta el 09 febrero 2019), en las líneas del identificador único que corresponden a SN está vacío. | PERMITIDO | |
|
|
PC SN Lot CAD |
08473543123678 687435268 L201JX32 Ene 2021 |
CASO 26: posición del Datamatrix a la derecha. | PERMITIDO |
2.2 ¿CÓMO EXPRESAR EL LOTE Y CADUCIDAD EN EL EMBALAJE EXTERIOR?: (Índice)
Se deben seguir las recomendaciones establecidas en el Apéndice IV del QRD (“Terms and abbreviations for batch number and expiry date to be used on the labelling of human medicinal products”). Este apéndice será actualizado próximamente para contemplar la posibilidad de utilizar la abreviatura ‘EXP’ en el embalaje exterior aunque ésta no sea la opción preferida por la AEMPS (cuando sea posible se deberá utilizar CAD).
En caso de utilizar en el etiquetado la abreviatura “EXP”, se deberá explicar su significado en el prospecto. Asimismo será aceptable utilizar la abreviatura ‘Lot’ en el cartonaje exterior, cuando sea necesario.
Adicionalmente, se considera aceptable a nivel nacional que Lot o Lote aparezca todo en mayúsculas: LOT o LOTE en el caso de que tecnológicamente no sea posible (p. ej. los inyectores en línea sólo puedan escribir en mayúscula).
2.3 MAQUETAS DE LOS FORMATOS NO COMERCIALIZADOS: (Índice)
La NI DS aplica a presentaciones comercializadas o cuya comercialización esté prevista antes del 9 de febrero de 2019 y exime a los restantes formatos.
Para los formatos comercializados, se acepta que se realice la solicitud tipo 19 “61(3)5 DS” independientemente de que se haya implementado efectivamente el IU (identificador único) y el DCM (dispositivo contra manipulaciones).
Para los medicamentos no comercializados antes del 9 de febrero de 2019 (medicamentos en suspensión temporal o autorizados antes de la NI DS y no comercializados aún), la solicitud tipo 19 “61(3) DS” se realizará en el momento que se produzca la comercialización.
2.4 ¿CÓMO SE INCORPORARÁ A LOS DATOS DEL REGISTRO EL GTIN DEL FORMATO CUANDO EL TAC OPTE POR ESTA ALTERNATIVA PARA IDENTIFICAR EL PC?: (Índice)
Se habilitará en la AEMPS un lugar donde el TAC pueda incluir el GTIN/NTIN en el caso de nuevos registros y en los medicamentos ya autorizados.
2.5 OFICIADO DE NOTIFICACIONES CONFORME AL ARTÍCULO 61(3): (Índice)
No se van a oficiar de forma específica las Notificaciones 61(3) Dispositivos de Seguridad.
2.6 EN RELACIÓN A LAS TASAS CUANDO SE TRATA DE UN MEDICAMENTO AUTORIZADO POR RM/DC: (Índice)
Las notificaciones del Artículo 61(3) europeas cuyo cambio sea única y exclusivamente la actualización de las secciones 17 y 18 para los procedimientos europeos (RM/DC) están EXENTAS DE TASA en España. La aplicación de RAEFAR II permite marcarlo al cargar la solicitud y así poder realizar el envío sin tasa.
2.7 CODIFICACIÓN DE LOS ELEMENTOS DEL IDENTIFICADOR ÚNICO: (Índice)
2.7.1 ¿Es obligatoria la inclusión del CN en el IU?:
Sí. El CN debe ir incluido entre los elementos del IU, bien formando parte del código de producto (PC) si se trata de un NTIN o bien como quinto elemento (posición 712 para España) en el caso de PC codificado como GTIN.
En el primer caso (NTIN), se recomienda NO incluir de forma repetida el CN como quinto elemento en la posición 712, por ser innecesario y poder llevar a confusión a los sistemas de lectura.
2.7.2 ¿Cómo debe ser el código de producto (PC) NTIN?:
La codificación del PC en forma de NTIN consiste en los 7 dígitos del CN (sin punto) precedidos siempre por los dígitos 0847000.
Ejemplo: 0847000XXXXXXX
2.7.3 Cuando se codifique el CN en el quinto elemento ¿cuántos dígitos, 6 ó 7, ha de tener el CN?:
El quinto elemento (posición 712 para España) debe tener los 7 dígitos del CN, sin punto.
2.7.4 Cuando se imprima el CN como NN ¿cuántos dígitos, 6 ó 7, ha de tener el CN?. En caso de que fueran 7, ¿el DC debe ir separado por un punto, tal y como figura en el embalaje exterior?:
De conformidad con lo establecido en la pregunta 2.7 del documento QA de la Comisión, y dado que en España ya es requisito la impresión del CN en el ángulo superior derecho de las caras principales del estuche, no es necesaria su inclusión de forma duplicada entre los elementos visibles del identificador único, por lo que no se recomienda su impresión.
No obstante, en caso de incluir el NN debe ser igual que el CN visible del embalaje exterior, es decir, con 6 dígitos, un punto y el 7º dígito de control.
2.7.5 En el caso de envases multilingües y que se requiera la inclusión del NN en el IU por decisión de las autoridades ¿cómo debe imprimirse cada NN?:
Para el NN de España (CN) deberán seguirse las indicaciones y recomendaciones expuestas en el punto anterior 2.7.4. En relación al NN correspondiente al otro Estado miembro se seguirán las indicaciones propuestas por las autoridades correspondientes.
En el caso específico de envases multilingües de España y Portugal, conforme a lo acordado con las autoridades portuguesas, se propone lo siguiente: el NN de Portugal debe imprimirse como “NN (PT)” y el NN de España puede no incluirse por estar ya impreso como CN en las caras principales (en caso de incluirse, de igual modo deberá aparecer como “NN (ES)”, si bien, tal y como se indica en el punto anterior no se recomienda su inclusión).
2.8 NECESIDAD DE INCLUIR EL BRAILLE EN 2 Ó 3 LÍNEAS Y ABREVIATURAS: (Índice)
En caso de que un medicamento tenga la información del braille y por motivos de espacio para incluir el IU se necesite ampliar a más líneas o a varias caras (p. ej. en la primera línea la marca del medicamento, en la segunda la dosis y en la tercera la forma farmacéutica): NO es necesario solicitar a la AEMPS variación IB. También será posible utilizar abreviaturas para la inclusión de las formas farmacéuticas (ver listado de abreviaturas habitualmente permitidas por la AEMPS ).
).
En cualquiera de los casos anteriores se deberá solicitar un nuevo certificado de Braille que se adjuntará junto con la Notificación 61(3) DS.
2.9 EN EL CASO DE MEDICAMENTOS QUE NO ESTUVIERAN AFECTADOS POR LA OBLIGACIÓN DE INCORPORAR DISPOSITIVOS DE SEGURIDAD, ¿DEBE ACTUALIZARSE LA PLANTILLA QRD INCLUYENDO LAS SECCIONES 17 Y 18 ANTES DEL 9 DE FEBRERO DE 2019?: (Índice)
No será necesario que la plantilla esté actualizada antes del 9 de Febrero de 2019 para aquellos medicamentos que no estén afectados por la obligación de incorporar dispositivos de seguridad.
En los medicamentos que no están afectados por el Reglamento Delegado de la Comisión, la actualización de la plantilla QRD se hará con motivo de una modificación que así lo requiera y se indicará en los epígrafes 17 y 18: “no procede”.
2.10 EN EL CASO DE MEDICAMENTOS QUE NO ESTUVIERAN AFECTADOS POR LA OBLIGACIÓN DE INCORPORAR DISPOSITIVOS DE SEGURIDAD PERO INCORPOREN UN CÓDIGO DE BARRAS BIDIMENSIONAL PARA OTROS OBJETIVOS DIFERENTES (P. EJ. PRECIO, REEMBOLSO, LOTE, CADUCIDAD…), ¿CÓMO DEBE NOTIFICARLO A LA AEMPS? (Índice)
Cuando se considere la inclusión de un código de barras bidimensional para otros objetivos diferentes a la identificación y verificación de la autenticidad del medicamento y no se incluya un IU (tal y como plantea la Pregunta 2.2 del documento QA de la Comisión), el TAC deberá presentar una variación de cambio de diseño (I)B C.I.z.
La inclusión de información adicional a través de un código quick response (código QR) seguirá las indicaciones establecidas en la nota informativa 27/2015 ‘Utilización de códigos quick response (QR) para proporcionar información sobre los medicamentos’.6
2.11 OTRAS CONSIDERACIONES A TENER EN CUENTA: (Índice)
No es necesario esperar a la Autorización de la Revalidación para que el TAC pueda implementar los DS. Adicionalmente, tendrá que realizarse la Notificación 61(3) DS.
No se considera aceptable la Notificación 61 (3) DS en el procedimiento conocido como “Shortened Renewal Procedure”.
No se permite agrupar variaciones de cambio de diseño de distintos medicamentos ya que no se ajusta al criterio de agrupación de variaciones.
Si una vez realizada la Notificación 61(3) DS hubiera que modificar la maqueta incluida en dicha Notificación y, siempre y cuando dicha modificación no afectase a la legibilidad, deberá presentarse la maqueta actualizada a través de la misma aplicación de RAEFAR II.
2.12 DISPOSITIVOS CONTRA MANIPULACIONES (DCM) (Índice)
Si la inclusión del DCM se realiza en medicamentos que carecen de embalaje exterior se deberá:
(1) Solicitar una modificación correspondiente al módulo 3 Calidad (3.2.P.7 Sistema del cierre del envase) y,
(2) realizar la Notificación 61(3) DS.
3. FABRICACIÓN Y CONTROL (Índice)
3.1 LA INSTALACIÓN DE EQUIPOS PARA LA SERIALIZACIÓN DE LOS ENVASES, ¿REQUIERE ALGÚN TIPO DE COMUNICACIÓN A LA AEMPS?: (Índice)
La instalación de los equipos de serialización en las líneas de acondicionado NO requiere autorización por parte de la AEMPS, por lo que no es necesario presentar ninguna solicitud de autorización. Podrá gestionarse a nivel interno con el control de cambios correspondiente, llevando a cabo las cualificaciones necesarias.
No obstante, si con motivo de la instalación de dichos equipos para la serialización se llevan a cabo otros cambios (ampliación de salas, traslado de líneas,…), si bien, con carácter general también se podrán llevar a cabo a nivel interno, se recomienda consultar con el Departamento de Inspección y Control de Medicamentos (sgicm@aemps.es) para valorar si fuera necesaria o no la autorización de los cambios. En cualquier caso, la adquisición de nuevas líneas de acondicionamiento siempre será objeto de autorización.
3.2 ETIQUETAS: (Índice)
El uso de etiquetas que contengan el IU sólo puede ser autorizado en determinadas circunstancias y siempre conforme a las condiciones señaladas en la respuesta a la pregunta 2.21 del documento QA de la Comisión.
Se permite la utilización de las etiquetas de determinados envases clínicos cuando dicha pegatina ya constituye el etiquetado del embalaje exterior que ya recoge toda la información, por lo que es coherente que también contenga la relativa al IU (pudiendo incluso ser dicha pegatina el dispositivo contra manipulaciones).
3.3 MUESTRAS GRATUITAS: (Índice)
Ver respuesta a la pregunta 8.5 del documento QA de la Comisión.
3.4 ¿CUÁLES SON LAS CONDICIONES QUE SE DEBEN CUMPLIR EN CASO DE QUE UN LABORATORIO ENCARGUE LA IMPRESIÓN DEL IU A UN TERCERO CONTRATADO?: (Índice)
El Reglamento delegado establece en su artículo 14 que “El fabricante que coloque los dispositivos de seguridad verificará que el código de barras bidimensional con el identificador único cumple lo dispuesto en los artículos 5 y 6, es legible y contiene la información correcta”.
En el caso de utilización de envases pre-impresos por una imprenta (sin autorización de laboratorio), se podría hacer una interpretación flexible de la pregunta 4.4 del documento QA de la Comisión, de forma excepcional y justificada, siempre que se cumplan las premisas descritas a continuación.
El laboratorio fabricante, en el proceso de acondicionamiento secundario de los medicamentos, es el que coloca estos dispositivos de seguridad en los medicamentos. En este sentido, el laboratorio deberá garantizar la verificación a la que hace referencia el artículo 14 (que el código de barras bidimensional con el identificador único cumple lo dispuesto en los artículos 5 y 6 del citado Reglamento, es legible y contiene la información correcta). Para esta verificación se deben cumplir las siguientes condiciones, además de las ya establecidas en la pregunta 4.4 del documento QA relativas al cumplimiento de Normas de Correcta Fabricación (NCF) de la actividad subcontratada (acuerdo técnico, procedimientos, auditorías…):
- La imprenta debe imprimir y verificar cada uno de los envases. La lectura para verificación tras la impresión es la condición necesaria para la creación del listado de IUs a trasladar por un medio seguro al fabricante.
- Idealmente, los envases ya impresos y verificados por la imprenta, deben ser verificados por el fabricante por un lector en línea. Esto tendría la ventaja de eliminar del listado los rechazos de la línea.
- Se podría aceptar que la verificación por el fabricante fuera de una muestra representativa. Para el muestreo habría que definir los criterios de aceptación requeridos. La verificación de las unidades muestreadas requeriría un lector, que permita, en su caso, también eliminar al menos las unidades rechazadas para destrucción, así como los envases sobrantes.
3.5 EN EL CASO DE UTILIZAR ENVASES PRE-SERIALIZADOS, ¿ES POSIBLE CARGAR EN EL REPOSITORIO TODOS LOS IU RECIBIDOS DEL PROVEEDOR QUE HA PRE-SERIALIZADO LOS ENVASES?: (Índice)
Según la pregunta 7.16 del documento QA de la Comisión, la carga en el repositorio de los IUs debe hacerse en un momento que permita que el sistema no se sobrecargue con información relativa a restos de fabricación, es decir, (partes de) lotes fabricados que no se han certificado.
En este sentido, se debe procurar en la medida de lo posible no cargar en el repositorio los IUs de las unidades rechazadas para destrucción, así como los envases sobrantes.
3.6 DISPOSITIVO CONTRA MANIPULACIONES (DCM): (Índice)
La integridad del DCM debe ser verificada por el farmacéutico antes de la dispensación del medicamento. Cualquier manipulación del DCM necesaria por parte del farmacéutico debe estar justificada y realizarse en presencia del paciente/usuario al que se le dispensa.
En el caso excepcional en el que se solapen DCM y cupón precinto, el farmacéutico podrá romper el DCM una vez haya verificado la integridad del DCM, pero en la medida de lo posible esta situación donde se solapan DCM y cupón precinto debe evitarse.
4. IMPORTACIONES PARALELAS: (Índice)
En España la distribución paralela de medicamentos de uso humano se regula según lo establecido en el Real Decreto 1785/2000, de 27 de octubre, sobre la circulación intracomunitaria de medicamentos de uso humano. En el artículo 2.4 del citado Real Decreto se permite el re-etiquetado mediante etiquetas autoadhesivas en el proceso de modificación de envasado de los medicamentos. Además, la Agencia Española de Medicamentos y Productos Sanitarios, en algunos casos, recomienda la elaboración de un nuevo cartonaje para los medicamentos de uso humano autorizados por importación paralela.
Por ello, para la colocación de los dispositivos de seguridad (identificador único-IU y dispositivo contra las manipulaciones-DCM) en los envases de los medicamentos de uso humano autorizados por importación paralela se debe tener en cuenta si el medicamento debe ser re-envasado o si se permite su re-etiquetado. En ambos casos, previamente a la colocación de los nuevos dispositivos de seguridad equivalentes, el importador paralelo debe verificar los dispositivos de seguridad del envase original y proceder a la desactivación del IU en el sistema de repositorios, conforme a lo establecido en el Reglamento delegado (UE) 2016/161.
4.1 PARA LOS MEDICAMENTOS QUE DEBEN SER RE-ENVASADOS: (Índice)
Para los medicamentos que deben ser re-envasados, el importador paralelo colocará los nuevos dispositivos de seguridad equivalentes en el nuevo envase. El IU será colocado preferentemente de forma impresa en el embalaje, tal y como establece el Reglamento delegado (UE) 2016/161, y sólo en las circunstancias establecidas en la pregunta 2.21 del documento de preguntas y respuestas de la Comisión, podrá autorizarse el uso de etiquetas y siempre que se cumplan todos los requisitos establecidos en dicha pregunta. No obstante, en caso de imposibilidad técnica para la impresión del IU en el envase, se recomienda utilizar envases con el IU pre-impreso siguiendo lo establecido en el punto 3.4 del documento de Recomendaciones para la aplicación de los dispositivos de seguridad de medicamentos de uso humano publicado en la web de la Agencia.
4.2 PARA LOS MEDICAMENTOS PARA LOS QUE SE PERMITE SU RE-ETIQUETADO: (Índice)
Para los medicamentos para los que se permite su re-etiquetado, el importador paralelo colocará los dispositivos de seguridad equivalentes cumpliendo con lo establecido en las preguntas 1.20, 1.21 y 1.22 del documento de preguntas y respuestas de la Comisión, en relación a sobre cómo proceder para la supresión de los dispositivos de seguridad del envase original, así como para la colocación de los nuevos dispositivos de seguridad equivalentes. En todo caso, las etiquetas utilizadas deben cumplir los requisitos recogidos en la pregunta 2.21 del documento de preguntas y respuestas de la Comisión.
El importador paralelo, de conformidad con la pregunta 1.20 del documento de preguntas y respuestas de la Comisión, debe demostrar que el nuevo DCM colocado en el envase puede ser considerado equivalente y es igualmente eficaz para proporcionar evidencia de manipulación como el DCM del envase original.
4.3 DISPOSITIVOS DE SEGURIDAD Y PRESTACIÓN FARMACÉUTICA: (Índice)
Los medicamentos de uso humano obligados a llevar dispositivos de seguridad y sujetos a prescripción médica que sean autorizados por importación paralela, deben mantener en el envase el código de barras del cupón precinto, hasta que la Dirección General de Cartera Básica de Servicios del Sistema Nacional de Salud y Farmacia del Ministerio de Sanidad, Consumo y Bienestar Social establezca disposiciones al respecto.
5. OPERACIONES DE LOS DISPOSITIVOS DE SEGURIDAD: (Índice)
5.1 MEDICAMENTOS EXTRANJEROS PARA ATENDER NECESIDADES ESPECIALES: (Índice)
Los medicamentos traídos conforme al artículo 5 de la Directiva 2001/83/CE NO están sujetos a las disposiciones del Reglamento Delegado, si bien la Comisión Europea recomienda que estos medicamentos sean verificados y desactivados antes de ser suministrados (pregunta 1.8 del documento QA de la Comisión).
En este sentido, la legislación nacional que se desarrolle en España incluirá esta disposición para estos medicamentos (siempre que les aplique); que sea verificada su autenticidad por la entidad que los importe y desactivados los IUs por la que los suministre.
5.2 SUMINISTRO DIRECTO DE ANESTÉSICOS LOCALES A PROFESIONALES SANITARIOS: (Índice)
Los anestésicos locales del grupo N01BB (incluidos en el Anexo de la Resolución de la Agencia Española de Medicamentos y Productos Sanitarios por la que se establece el listado de medicamentos que pueden venderse directamente a los profesionales de la medicina, odontología y podología para el ejercicio de su actividad profesional y de la Resolución de la Agencia Española de Medicamentos y Productos Sanitarios por la que se establece el listado de medicamentos de uso humano que pueden venderse directamente a los profesionales de la veterinaria para el ejercicio de su actividad profesional) podrán venderse directamente desde los laboratorios o entidades de distribución a los citados profesionales, y corresponderá a los laboratorios o entidades de distribución cumplir con las obligaciones derivadas del Reglamento delegado (UE) 2016/161 de la Comisión, debiendo verificar los dispositivos de seguridad y desactivar el identificador único (IU) de los medicamentos que suministren a dichos profesionales.
Sin perjuicio de que lo posteriormente se detalle en la legislación nacional que se desarrolle, esta obligación para los laboratorios o entidades de distribución con respecto a las operaciones con los dispositivos de seguridad ya viene recogida en la disposición adicional tercera de venta directa a profesionales sanitarios del Real Decreto 782/2013, de 11 de octubre, sobre distribución de medicamentos de uso humano, por lo que es de aplicación desde el 9 de febrero de 2019.
6. MEDIDAS PARA DISMINUIR EL NÚMERO DE ALERTAS: (Índice)
6.1 CARACTERES DEL NÚMERO DE SERIE Y LOTE: (Índice)
Tras la puesta en marcha del sistema se ha observado que se han generado numerosas alertas en el sistema con motivo de la lectura errónea por parte de los lectores de los usuarios finales de determinados caracteres incluidos en la codificación del número de serie y lote.
Es por ello que, con el objeto de evitar estas numerosas alertas innecesarias, que dependen de las posibles configuraciones de los lectores, se recomienda a los laboratorios fabricantes que colocan los dispositivos de seguridad sobre el envase de los medicamentos de uso humano que acoten el estándar GS1 para la codificación del número de serie y lote a los siguientes requisitos:
- Uso exclusivo de mayúsculas.
- No utilización de caracteres especiales (ej. guiones, barras…).
- Evitar el uso de las letras “Y” y “Z” (afecta a otros países).
Referencias
- La AEMPS recomienda no incluir los paréntesis que indican la posición en el datamatrix puesto que puede inducir a error cuando sea necesario introducir los datos de forma manual (en este caso no debe incluirse la numeración entre paréntesis).
- Ver pregunta 2.10 del documento QA de la Comisión.
- Según lo previsto en la pregunta 2.17 del documento QA de la Comisión, sería aceptable si bien se recomienda la agrupación PC/SN y Lote/CAD.
- Ver pregunta 2.18 del documento QA de la Comisión Europea. En este caso se agrupa toda la posible casuística.
- Notificaciones según el art. 61(3) de la Directiva 2001/83/CE
- Nota informativa 27/2015 ‘Utilización de códigos quick response (QR) para proporcionar información sobre los medicamentos’. https://www.aemps.gob.es/informa/notasInformativas/industria/2015/docs/NI-MUH_27-2015-codigos-quick-response.pdf
Si desea localizar información relacionada con el contenido de esta página, utilice el buscador