 Directrices para la resolución de variaciones de adecuación al RD 1345/2007 y otras variaciones coexistentes en el tiempo
Directrices para la resolución de variaciones de adecuación al RD 1345/2007 y otras variaciones coexistentes en el tiempo
- Compartir:
-

-

-

-

Última actualización: 21/05/2010
DIRECTRICES PARA LA RESOLUCIÓN DE VARIACIONES DE ADECUACIÓN
AL RD 1345/2007
Y OTRAS VARIACIONES COEXISTENTES EN EL TIEMPO
21 de mayo de 2010
Antecedentes
La Disposición transitoria tercera del
Real Decreto 1345/2007, de 11 de octubre , por el que se regula el
procedimiento de autorización, registro y condiciones de dispensación de los
medicamentos de uso humano fabricados industrialmente, regula la solicitud de
modificaciones al etiquetado y prospecto de los medicamentos por parte del
titular de una autorización de comercialización (TAC) para ajustarse a las
exigencias que establece dicho RD.
, por el que se regula el
procedimiento de autorización, registro y condiciones de dispensación de los
medicamentos de uso humano fabricados industrialmente, regula la solicitud de
modificaciones al etiquetado y prospecto de los medicamentos por parte del
titular de una autorización de comercialización (TAC) para ajustarse a las
exigencias que establece dicho RD.
Estas solicitudes, no obstante, pueden coincidir en el tiempo con otros procedimientos en curso susceptibles de afectar al contenido del etiquetado y prospecto. Por ello, y con el fin de incrementar la eficiencia, evitar duplicidades y facilitar la labor de los solicitantes, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha elaborado estas directrices que permiten sistematizar la documentación necesaria a presentar por los titulares e informan de los criterios que, en cada supuesto resultarán de aplicación.
¿Qué se entiende por adaptación al RD 1345/2007?
En las solicitudes de modificación de adecuación a las exigencias del RD en ficha técnica, prospecto y etiquetado, la AEMPS realizará la evaluación de los siguientes aspectos de la información del medicamento:
-
Nombre del medicamento (denominación + dosis + forma
farmacéutica). La expresión de la dosis deberá ajustarse a las nuevas
recomendaciones del grupo Quality Review of Documents Group (QRD),
“QRD
Recommendations on the Expression of Strength in the Name of Centrally
Authorised Human Medicinal Products”
.
-
Composición:
- Adaptación de la composición a la expresión de la dosis que aparezca en el nombre.
- Denominación de los excipientes por su DOE o Farmacopea (según corresponda).
- Inclusión del número E (en su caso).
- Inclusión cuali y cuantitativa de los excipientes de declaración obligatoria en el apartado 2 de la ficha técnica (FT).
- Forma farmacéutica, adecuada a los Términos estándar de la EDQM (referencia).
-
Excipientes de declaración obligatoria y texto
correspondiente de la
circular de la AEMPS Nº 2/2008: Información sobre los excipientes en el
etiquetado, prospecto y ficha técnica de los medicamentos de uso
humano.
- En Etiquetado, además:
- Incorporación o exclusión de los símbolos, siglas y leyendas correspondientes (entre ellos, cuando proceda y a petición de la compañía, las siglas EFP).
- Tiempo de validez del medicamento tras su apertura, dilución o reconstitución e inclusión del recuadro para consignar la fecha de apertura, dilución o reconstitución.
- Inclusión de la indicación terapéutica (que aparece en el prospecto) en el etiquetado de los medicamentos no sujetos a prescripción médica.
- Inclusión del representante local, si lo tuviera.
- Supresión del comercializador.
- Inclusión de un recuadro o espacio en blanco (sin barniz) para indicar la posología recetada y duración del tratamiento.
- Nombre en Braille en el embalaje exterior en los casos que proceda.
- Test de legibilidad. Se aceptará la solicitud de exención de test de legibilidad en aquellos productos que hayan sido o estén siendo sometidos a un arbitraje y en los casos aceptados por el grupo europeo CMDh. Los casos en los que las solicitudes de adecuación se hubieran presentado incluyendo un test de legibilidad sobre textos no autorizados serán estudiados uno a uno, minimizando la realización de nuevos test en la medida de lo posible a través de la realización de informes puente.
RESOLUCIÓN DE LAS VARIACIONES RELACIONADAS CON LA ADAPTACIÓN AL RD 1345/2007
La clasificación de solicitudes de modificación se establece en el siguiente árbol de decisión
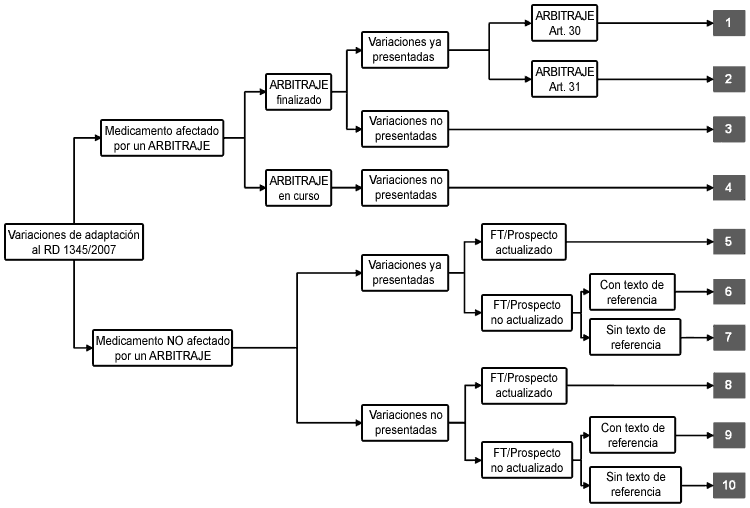
1. Variaciones ya presentadas y que afectan a un medicamento que tiene un arbitraje ya finalizado (articulo 30 de la Directiva 2001/83/CE y modificaciones) (1)
Como norma general, las variaciones de adaptación a RD 1345/2007 o de modificación de fichas técnicas presentadas, se resolverán junto con el arbitraje una vez que las compañías hayan solicitado la correspondiente variación.
- Se realizará una consulta en RAEFAR que permita conocer las variaciones en trámite de los medicamentos afectados por un arbitraje artículo 30.
- Las variaciones pendientes de Clínica y Farmacovigilancia referidas a aspectos evaluados en el procedimiento de arbitraje (FT y Prospecto), quedarán resueltas junto con la variación de implementación la Decisión de la Comisión (variación IB46).
- Si hubiera alguna variación de adecuación al RD 1345/2007 del principio activo objeto del arbitraje, la evaluación de la AEMPS respecto a estas variaciones se limitará al etiquetado. Se aceptará la solicitud de exención de presentación del test de legibilidad en aquellos medicamentos que hayan sido o estén siendo sometidos a un procedimiento de arbitraje.
2. Variaciones ya presentadas y que afectan a un medicamento que tiene un arbitraje ya finalizado (articulo 31 de la Directiva 2001/83/CE y modificaciones) (2)
Por lo general, en los arbitrajes por el artículo 31, la armonización se reduce a puntos específicos de la FT. No obstante, en algunos casos, el Comité de Medicamentos de Uso Humano (CHMP) realiza una armonización completa de la FT, etiquetado y prospecto. En esta segunda situación, las modificaciones pendientes se resolverían según el procedimiento descrito en el punto 1. En el caso más general, que es un Art 31 sin armonización de FT y prospecto, para medicamentos autorizados por Procedimiento Nacional y que tengan FT autorizada se incluirá el resultado del arbitraje art. 31 en el punto específico de la FT. Para medicamentos autorizados por Procedimiento Nacional y que no tengan FT autorizada deberá solicitar además la variación correspondiente. En el caso de estar pendiente una variación de solicitud de FT, ambas modificaciones se resolverían simultáneamente.
3. Variaciones NO presentadas y que afectan a un medicamento que tiene un arbitraje ya finalizado
Como norma general, si existe un arbitraje ya finalizado y hubiera la obligación por parte de la compañía de presentar variaciones de adaptación a RD 1345/2007 o modificación de ficha técnica, estas se resolverán -de acuerdo con el punto 1 del presente documento- presentando las siguientes variaciones:
- Variación de adaptación a RD para los materiales y etiquetado (tipo II.500 según la legislación vigente; Tipo IB según el nuevo Reglamento).
- Variación de implementación del arbitraje para resolver ficha técnica y
prospecto de acuerdo a éste
(Tipo IB según la legislación vigente; tipo IAin/IB/II según el nuevo reglamento; ver pie de pagina nº 2, página 2 del presente documento).
4. Variaciones NO presentadas y que afectan a un medicamento que tiene un arbitraje en marcha
Si hubiera la obligación por parte de la compañía de presentar variaciones de adaptación a RD 1345/2007 o modificación de ficha técnica y existe un arbitraje en marcha, podrá optarse por esperar a la resolución de dicho arbitraje para actuar de acuerdo con el punto 1.
En el caso de tener que presentar la revalidación quinquenal, el TAC deberá solicitar la adecuación a RD 1345/2007 (etiquetado y ficha técnica y prospecto si existen textos actualizados3 en paralelo, según lo establecido en las disposiciones de dicho RD. Sin embargo, al estar el producto sometido a un arbitraje, se exime al TAC de la responsabilidad de presentar el test de legibilidad. Para ello, deberá presentarse la solicitud de exención correspondiente en la documentación aportada con la renovación.
En los casos en los que, según lo establecido en la disposición transitoria tercera del RD 1345/2007, el TAC este obligado a presentar la adecuación al RD junto con cualquier variación tipo II (exceptuando las que afecten a la calidad del medicamento), en lo referido al test de legibilidad se procederá de la misma manera que con las renovaciones.
5. Variaciones de adaptación a RD ya presentadas con FT o prospecto actualizado y sin arbitraje finalizado o en marcha
En este caso la AEMPS resolverá todo lo relacionado con la variación de adaptación a RD. (Variación tipo II-500 según la legislación vigente y Tipo IB según el nuevo reglamento).
6. Variaciones de adaptación a RD ya presentadas con FT o prospecto NO actualizado, sin arbitraje finalizado o en marcha, con un texto de referencia armonizado
En este caso la AEMPS resolverá la adecuación a RD (etiquetado, ficha técnica y prospecto) y la actualización de la FT siempre y cuando se den las siguientes condiciones:
- Que exista un texto (ficha técnica y prospecto) reciente que se considere adecuado. La Agencia remitirá al TAC dicho texto de referencia armonizado para su implementación; y
- Que el texto remitido por el TAC sea idéntico al proporcionado por la Agencia salvo en aquellos aspectos que inevitablemente deban diferir.
El resultado será la adaptación a RD y la actualización de la información de medicamento a un texto de referencia armonizado. Se aceptará la solicitud de exención de realización de los test de legibilidad en los casos acordados por el grupo CMDh.
7. Variaciones de adaptación a RD ya presentadas con FT o prospecto NO actualizado, sin arbitraje finalizado o en marcha, SIN un texto de referencia armonizado
En este caso la AEMPS resolverá todo lo relacionado con la variación de adaptación a RD (ficha técnica, prospecto, etiquetado, y materiales), pero no se resolverá la adecuación de la FT y prospecto a QRD, por lo que seguirá existiendo la obligación por parte del titular de presentar –en caso de que no se hubiere hecho- una variación para actualizar ficha técnica y prospecto (Variación tipo II-500 según la legislación vigente y Tipo II según el nuevo reglamento).
8. Variaciones de adaptación a RD NO presentadas a fecha de publicación de este documento con FT o prospecto actualizado y sin arbitraje finalizado o en marcha
En este caso se presentará la variación de adaptación a RD según lo establecido en la Disposición Transitoria tercera del RD 1345/2007 (Variación tipo II-500 según la legislación vigente y Tipo IB según el nuevo reglamento).
9. Variaciones de adaptación a RD NO presentadas a fecha de publicación de este documento con FT o prospecto NO actualizado, sin arbitraje finalizado o en marcha, y con un texto de referencia armonizado
En este caso el TAC deberá presentar la variación de actualización de ficha técnica correspondiente. (Variación tipo II-500 según la legislación vigente y Tipo II según el nuevo reglamento).
Según lo establecido en la disposición transitoria tercera del RD 1345/07, el TAC deberá presentar la adecuación a RD junto con la solicitud de actualización de FT y ambos aspectos se resolverán en la misma variación.
10. Variaciones de adaptación a RD NO presentadas a fecha de publicación de este documento con FT o prospecto NO actualizado, sin arbitraje finalizado o en marcha, y SIN un texto de referencia armonizado
En este caso el TAC deberá presentar la variación de actualización de ficha técnica correspondiente. (Variación tipo II-500 según la legislación vigente y Tipo II según el nuevo reglamento).
Según lo establecido en la disposición tercera del RD 1345/07, el TAC deberá presentar la adecuación a RD junto con la solicitud de actualización de FT y ambos aspectos se resolverán en la misma variación.
1 Arbitrajes con armonización de la FT, a nivel europeo de un medicamento de referencia. Independientemente del procedimiento de autorización (Nacional/RM/DC); -el contenido del arbitraje se aplica a las licencias- y genéricos (no cubiertos por el arbitraje).
2 Aplicación del Reglamento CE/1084/2003 para los arbitrajes de medicamentos autorizados por Procedimiento Nacional, presentados con anterioridad a la fecha de publicación de la modificación del R.D. 1345/2007, solicitando una modificación Tipo IB46 al titular(es). A partir de la publicación de dicha modificación del RD, se aplicará el nuevo Reglamento de modificaciones CE 1234/2008 de acuerdo a: (a) modificación Tipo IAIN en medicamentos cubiertos por el arbitraje (se implementará directamente la FT resultante del arbitraje aunque el medicamento al que se aplique tenga menos información que la FT resultante del arbitraje); (b) modificación Tipo IB para medicamentos NO cubiertos por el arbitraje, cuando la FT resultante del arbitraje SI es igual a la FT del medicamento NO incluido en el arbitraje y la implementación del cambio NO supone la inclusión de información adicional (aplica a genéricos y licencias del medicamento de referencia); y (c) una modificación Tipo II para medicamentos NO cubiertos por el arbitraje, cuando la FT resultante del arbitraje NO es igual a la FT del medicamento NO incluido en el arbitraje y la implementación del cambio SI supone la inclusión de información adicional (aplica a genéricos y licencias del medicamento de referencia).
2 Arbitrajes por principio activo, por grupo o clase farmacológica por motivos de seguridad o eficacia. En este caso dentro del arbitraje estarán incluidos los medicamentos de referencia, licencias y genéricos.
3 A los efectos del presente documento, se entenderán por textos actualizados y/o textos de referencia aquellos de medicamentos con igual composición en principios activos y misma vía de administración que provengan de arbitrajes, o aquellos revisados e implementados por la AEMPS en los dos últimos años excluyendo variaciones menores, solo de calidad y aspectos administrativos.
Si desea localizar información relacionada con el contenido de esta página, utilice el buscador